|
Biochemistry and Cell Biology 76, 129-137.
| Abstract: Much of the fruit fly genome is compact ("E. coli mode"), indicating a genome-wide selection pressure against DNA with little adaptive function. However, in the bithorax complex (BX-C) homeodomain genes are widely dispersed with large introns ("mammalian mode"). Chargaff difference analysis of compact bacterial and viral genomes has shown that most mRNAs have the potential to form stem-loop structures with purine-rich loops. Thus, for many taxa if transcription is to the right, the "top" (mRNA synonymous) DNA strand has purine-rich loop potential, and if transcription is to the left the "top" (template) strand has pyrimidine-rich loop potential. The best indicator bases for transcription direction are A and T for AT-rich genomes, and C and G for CG-rich genomes. Consistent with this, Chargaff difference analysis of BX-C genes and several non-BXC genes shows that, whatever the mode, mRNAs have the potential to form stem-loop structures with A-rich loops. We confirm that many potential open reading frames (ORFs) in the BX-C are unlikely to be functional. Conversely, we suggest that a few unassigned ORFs may actually be functional. Since apparent organization in the mammalian mode cannot be explained in terms of unacknowledged ORFs, yet the fruit fly genome is under pressure to be compact, it is likely that many BX-C functions do not involve the encoding of proteins. |
Introduction
Transcribed duplex DNA has an mRNA-synonymous strand and a template strand. If transcription is to the right the top strand is the mRNA-synonymous strand. If transcription is to the left the top strand is the template strand. Three decades ago Szybalski et al. (1966) showed that mRNA-synonymous strands have purine-rich clusters, and Chargaff et al. showed that, to a close approximation, Chargaff's first parity rule for duplex DNA (%A=%T and %C=%G; Chargaff, 1951), also applies to single-stranded DNA ("Chargaff' second parity rule"; Karkas et al. 1968; Chargaff, 1979). The second parity rule has become particularly apparent as the results of various genome sequencing projects have emerged (Prabhu, 1993). For example, the "top" strand of chromosome III of Saccharomyces cerevisiae (Oliver et al. 1992), has 98212 As, 95572 Ts, 62125 Cs, and 59432 Gs. A and T differ by only 2640 bases, and C and G differ by only 2693 bases. Only 1.4% of the W bases (A and T, which pair weakly) are not accounted for by a potential pairing partner. Only 2.2% of the S bases (C and G, which pair strongly) are not accounted for by a potential pairing partner. The second parity rule holds not only for the entire sequence, but also for small windows (e.g. 1 kb) in the sequence, albeit usually with less precision.
Combining Szybalski's observation with Chargaff's second rule, it follows for mRNA-synonymous strands, either that purines in the clusters are balanced by an equal number of dispersed pyrimidines, or that there are small deviations from the second rule in favour of purines. That such deviation are present, and can act as indicators of transcription direction, is apparent from the original observations on bacterial and bacteriophage genomes by Szybalski et al. (1966), by further bacterial data from the Chargaff laboratory (Rudner et al. 1969), by a study of various mammalian genes and viruses by Smithies et al. (1981), and by studies from our laboratory (Forsdyke and Bell, 1997).
For example, Smithies et al. (1981) noted that the "top", mRNA-synonymous, strand of rightward-transcribing globin genes has negative "Chargaff differences", when assessed as C-G (i.e. G>C). In the circular SV40 virus genome they noted that negative Chargaff differences in the top strand (i.e. G>C) correlate with the rightward-transcribing late genes (in which the bottom strand is the template strand), and positive Chargaff differences in the top strand (i.e. C>G) correlate with the leftward-transcribing early genes (in which the top strand is the template strand). Thus, as shown in the prescient Fig. 4 of Szybalski et al. (1966), for the "top" strand of DNA, leftward transcription is indicated by pyrimidine excess, and rightward transcription is indicted by purine excess. Deviations from Chargaff's second parity rule (Chargaff differences) are found in transcribed regions, usually associated with purine-rich clusters in the mRNA-synonymous strand, and complementary pyrimidine-rich clusters in the template strand (Szybalski et al. 1966).
The generality of this is further suggested by the observation of Mrazek and Kypr (1994) that A>T universally in the mRNA-synonymous strand of protein-encoding sequences (from bacteria to primates). From their data on 3,954 primate coding sequences, the average Chargaff difference for the W bases (calculated as (A-T)/W and expressed as a percentage), is 8.4%. Similarly, the base composition of 1657 human coding sequences compiled by Karlin and Mrazek (1996), yields a generic Chargaff difference (calculated as [R-Y]/[R+Y] and expressed as a percentage) of 4%. Usually, one Watson-Crick base pair serves as the main indicator of transcription direction. In CG-rich genomes or genome sectors, strands which are transcribed to the right are usually G-rich (G>C); in AT-rich genomes or genome sectors, strands which are transcribed to the right are usually A-rich (A>T).
To evaluate its power as an analytical tool in the characterization of genomes, we here apply "Chargaff difference analysis" to the bithorax complex (BX-C) and other genes in the AT-rich genome of the fruit fly Drosophila melanogaster. The sequence of BX-C has many unusual features (Martin et al. 1995; Lewis et al. 1995).
| While much of the genome is compact ("E. coli mode"), the BX-C appears to have escaped a genome-wide selection pressure against DNA with little adaptive function (Petrov et al. 1996). The BX-C is organized like mammalian genomes with much intergenic DNA and large introns ("mammalian mode"). The three homeodomain-containing proteins of the BX-C (ABD-A, ABD-B, UBX) are encoded by only 1.4% of the 315 kb sequence. There are at least two RNA-encoding genes (iab-4, bxd), many open reading frames (ORFs) considered unlikely to be functional, and many A-rich (A>T) and T-rich (T>A) clusters. Genetic studies have identified 12 cis-regulatory regions influencing development (Lewis, 1992), but their relationship to known sequence features is uncertain. A 338 kb segment from the 89E region of the third chromosome of Drosophila melanogaster (Martin et al. 1995) contains the 315 kb BX-C flanked by apparently unrelated regions in which genes are compactly arranged in the fashion of the rest of the genome. Thus one sequence segment contains contiguous genomic regions organized both in E. coli and in mammalian modes. |
We show here that in both regions transcription direction correlates with enrichment for the appropriate W base (A>T when transcription is to the right; T>A when transcription is to the left). Furthermore, Chargaff difference analysis supports the designation of many potential ORFs as non-functional.
| Since apparent organization in the mammalian mode cannot be easily explained by unacknowledged ORFs, yet the fruit fly genome is under pressure to be compact (Petrov et al. 1996), our results are consistent with the view that many of the genetic functions of the BX-C do not involve the encoding of proteins (Lewis et al. 1995). |
Methods ![]()
Chargaff difference analysis
Programs of the Genetics Computer Group (Gribskov and Devereux, 1991) were used to determine base composition (WINDOW), and, when appropriate, to randomize base order (SHUFFLE). Windows (1 kb) were moved along the sequence in steps of 100 nucleotides, and base compositions were determined in each window. A, C, G, and T refer to the quantity of the corresponding base in each window. Chargaff differences for the W bases were calculated either as (A-T)/W, or as (T-A)/W, or (for absolute values) as DW/W. These values were expressed as percentages. W is the sum of the W bases (W=A+T), and DW is the absolute value of the difference between the number of W bases (DW=|A-T|). Chargaff differences for the S bases were calculated either as (C-G)/S, or (for absolute values) as DS/S, and expressed as percentages. S is the sum of the S bases (S=C+G), and DS is the absolute value of the difference between the number of S bases (DS=|C-G|). Statistical analyses were as described by Meyer and Krueger (1994).
Results ![]()
A>T when transcription is to the right; T>A when transcription is to the left.
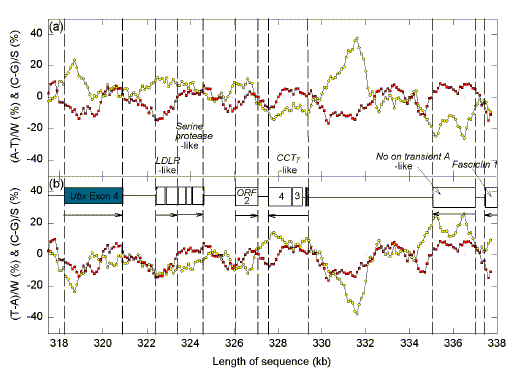
A 1 kb window was moved in 100 nt steps along the 338,324 nt sequence containing the BX-C. From the base composition of each window Chargaff differences for the W bases (A and T), and for the S bases (C and G) were calculated separately. The values were compared with known sequence features as documented in the GenBank entry and the corresponding papers (Martin et al. 1995; Lewis et al. 1995). Since much of the BX-C is uncharted in terms of sequence features, we demonstrate our approach by considering the fourth exon of the Ubx gene at the centromeric end of the complex and the adjoining "normal" DNA with more compactly arranged genes. Chargaff differences for the W bases are expressed either as (A-T)/W (Fig. 1a), or as (T-A)/W (Fig. 1b), whereas Chargaff differences for the S bases are expressed as (C-G)/S. There are several genes in the 20 kb region shown, whereas upstream of the fourth exon of Ubx (open box at the left) the BX-C contains a 51 kb intron (not shown), with relatively few acknowledged sequence features (although anterobithorax and bithorax map here).
|
|
| Fig. 1. Chargaff difference analysis of the centromeric flank of the BX-C. Variation of Chargaff differences are shown for a 20 kb segment at the centromeric end of the BX-C. A 1 kb sequence window was moved in steps of 100 nt and base compositions were determined in each window. From these values Chargaff differences were calculated independently for the W bases (grey circles) and for the S bases (black squares). Values for the W bases were calculated in (a) as (A-T)/W, and in (b) as (T-A)/W. Values for the S bases were calculated as (C-G)/W. Data points are located at the centre of each window. Exons and ORFs (hypothetical and proven) are shown as open boxes. Vertical dashed lines correspond to the limits of exons or ORFs. Horizontal arrows indicated the transcription direction assigned to each ORF. For further details see legend to Table 1. |
Moving from left to right, it is noted in Fig. 1b that Chargaff differences for the W bases are negative in the region of the rightwardly transcribed Ubx exon 4 (i.e. A>T). The BX-C then ends (between nts 320,921 and 322,431), and a set of five flanking ORFs are encountered [encoding LDL receptor-like repeats (orfld, orfla), and a serine protease-like protein (ORFS 331, 332, 333)]. These are also rightwardly transcribed and again A>T. There is then a space in which Chargaff difference values for the W bases become weakly positive (T>A in Fig. 1b) before the rightwardly transcribed ORF-2 is encountered. Here the sequence again becomes relatively A-rich.
The four exons of the CCTg-like gene (encoding a protein similar to the chaperonin-containing t-complex protein-1g subunit) are then encountered. These exons are numbered in decreasing order, since transcription is from the left. Here Chargaff difference values for the W bases (Fig. 1b) become positive (T>A). Further to the right, in the 2 kb region around nucleotides 330,000 and 332,000, the values become very negative (A>T). If there were an ORF here it would be transcribed to the right (see later). Chargaff differences for the W bases then fluctuate close to zero until two leftward transcribed genes are encountered after nucleotide 335,000. Here again the region becomes relatively T-rich (T>A). Thus all the ORFs recognized in the region conform to the rule that the upper strand is relatively A rich when transcription is to the right, and relatively T rich when transcription is to the left.
In keeping with the W-richness of the Drosophila genome, the W bases, rather than the S bases, appear important for predicting transcription direction. However, Chargaff differences for the less reliable Watson-Crick base pair predictor (the S bases in the present case) are still of considerable value, since regions where lines for the S bases and for the W bases cross may correspond to the limits of transcription units; furthermore, Chargaff differences for both the W and the S bases tend to be greatest in the middle of transcription units (Forsdyke and Bell, 1997; Bell and Forsdyke, 1999a,b). Sometimes these features are most easily seen when Chargaff differences for the W bases are expressed as (A-T)/W (Fig. 1a), and sometimes when Chargaff differences for the W bases are expressed as (T-A)/W (Fig. 1b). There is some suggestion that Chargaff differences are greatest near the middle of transcription units in the cases of the leftward transcribing CCTFg -like gene, and the no-on transient A-like gene (Fig. 1a).
| Transcription to the right | ||||||
| . | W bases | S bases | ||||
| . | (T-A)/W (%) | . | (C-G)/S (%) | |||
| Gene or ORF | Exon # | Natural | Shuffled | Exon # |
Natural | Shuffled |
| AHCY | 7 (3) | 2.3 | -2.1 | 7 (6) | -4.9 | 2.9 |
| a-Actinin-3 | 1 (1) | -12.5 | -0.3 | 1 (1) | -6.3 | 3.5 |
| AbdB | 8a(7) | -14.5 | -2.2 | 8a(1) | 7.1 | 2.4 |
| abdA | 7 (6) | -11.9 | -1.4 | 7 (2) | 9.5 | 0.3 |
| bxd | 8b(5) | -2.4 | -0.1 | 8b(5) | 1.0 | 3.6 |
| Ubx | 4 (2) | -12.3 | 1.4 | 4 (1) | 1.2 | 1.0 |
| LDLR | 2 (2) | -10.3 | 3.0 | 2 (2) | -8.6 | 0.005 |
| Ser. protease | 3 (3) | -6.3 | 0.6 | 3 (0) | 3.3 | -4.9 |
| ORF-2 | 1 (1) | -6.7 | -1.24 | 1 (0) | -0.6 | 4.9 |
| Average of 9 genes | -8.30�1.84 | -0.27�0.58 | . | -0.18�2.01 | 1.53�0.98 | |
| Probability not different from zero | P=0.002 | P=0.66 | . | P=0.93 | P=0.16 | |
| Probability no |Natural|>|Shuffled| | P=0.004 | . | . | P=0.60 | . | |
| Transcription to the left | ||||||
| gene 5 | 8c(4) | 1.7 | -1.4 | 8c(5) | 0.2 | 0.4 |
| iab-4 | 2 (0) | -1.9 | 0.7 | 2 (2) | 3.9 | 3.4 |
| glut-l | 1 (0) | -14.3 | 1.8 | 1 (1) | 5.5 | -1.1 |
| CCTg | 3d(3) | 9.1 | 3.5 | 3 (2) | 1.2 | -0.6 |
| no-on trans. | 1 (1) | 17.3 | 2.9 | 1 (1) | 6.2 | 2.3 |
| fasciclin I | 1 (1) | 6.6 | -3.8 | 1 (0) | -11.6 | 6.4 |
| Average of 6 genes | 3.10�4.39 | 0.61�1.13 | . | 0.91�2.69 | 1.81�1.15 | |
| Probability not different from zero | P=0.51 | P=0.61 | . | P=0.75 | P=0.18 | |
| Probability not |Natural|>|Shuffled| | P=0.60 | . | . | P=0.81 | . | |
| Note: Chargaff
difference values for windows (1 kb) whose centres overlap exons or ORFs (see Fig. 1) were
averaged to obtain values for exons or ORFs in the corresponding genes. Averages for all
genes are shown with standard errors of the mean. The total number of exons associated with each gene are listed in the columns headed "Exon #", together (in parentheses) with the number of exons which, in the natural sequence, conform to the Chargaff difference rule (R>Y if transcription is to the right, Y>R if transcription is to the left). Since the BXC region of chromosome 3 of Drosophila melanogaster is AT-rich (C+G= 41.7%), the W bases are expected to indicate transcription direction. Genes are listed as ordered in the chromosome (telomeric -> centromeric). Abbreviations: AHCY, putative homolog of the human gene encoding S-adenosylhomocysteine hydrolase; a-Actinin 3 = ORF similar to human a-Actinin 3; Abd-B, abd-A, and Ubx are genes encoding homeodomain proteins; bxd, and iab-4 are BX-C genes encoding an RNA product; LDLR, putative homolog of human gene encoding LDL receptor-like repeats; Ser. protease, putative homolog of gene encoding serine protease; ORF-2, open reading frame number 2; gene-5, candidate gene number 5; glut-l, putative homolog of human GLUT3 gene encoding a human glucose transporter type 3; CCTg, gene encoding putative homolog of the mammalian chaperonin-containing t-complex polypeptide g-subunit; no-on trans., ORF with similarity to no-on transient A gene; fasciclin I = last exon of gene encoding fasciclin I. a Exon 2 of Abd-B did not include the centre of a window, so was excluded.b Exon 6 of bxd overlaps exon 5 and was excluded. c Exon 4 of candidate gene 5 did not include the centre of a window, so was excluded. d Exon 1 of CCTg did not include the centre of a window, so was excluded. |
||||||
Each data point in Fig. 1 corresponds to the centre of a 1 kb window. Where data points overlap exons or ORFs, the values can be summed and averaged to obtain mean Chargaff difference values corresponding to the W and S bases for individual exons or ORFS, and the corresponding genes. Rightward and leftward transcribed exons, ORFs and genes are listed separately in Table 1.
It is noted that, with the notable exception of the AHCY gene in the telomeric flank of the BX-C, A>T for most rightward-transcribed exons or ORFs, including 5 out of the 8 exons of the bxd gene (which is thought to have an RNA, not a protein, product). T>A for most exons or ORFs which are leftward transcribed, with the notable exceptions of another gene with no protein product (iab-4), and a putative glucose transporter ORF (GLUT3). Average Chargaff difference values for rightward transcribed genes are significantly biased in the case of the W bases (A>T; P=0.002), but this is not found in the case of the S bases; for leftward transcribed genes, no significant biases are noted. The analysis was repeated with a randomized version of the sequence in which the bases had been shuffled to disrupt base order, while still maintaining overall base composition (Table 1). The same sequence positions were analyzed, but no consistent pattern of differences was observed. We believe the exceptions shown in Table 1 are special cases, for which explanations should be found (see Discussion).
The applicability of the rule to Drosophila was further supported by analysis of natural and shuffled versions of various genomic sequences selected as the first relatively long (approx. 10 kb) fruit fly sequences we happened to encounter in a GenBank search (Table 2). In these cases definition of the "top" strand was arbitrary and transcription was to the right. In this series, although A>T was still the most significant indicator (in keeping with the general AT-richness of the genome), there was also a significant excess of Cs (C>G).
Unassigned ORFs do not explain why BX-C has mammalian mode organization.
In their initial analysis of the 338 kb segment containing the BX-C, Martin et al. (1995) identified 60 ORFs showing preferential usage of Drosophila codons, and an additional 15 with low codon preference, but exceptional size (>200 amino acids); some of these were reported as likely to be functional based on similarities with known proteins, and some were rejected as likely to be non-functional. To obtain further evidence that some ORFs appearing in the long, apparently featureless, regions of the BX-C, are not functional, we calculated Chargaff difference values for the rejected ORFs whose coordinates were kindly provided by S. Celniker.
Table 3 shows a list of the ORFs and corresponding Chargaff differences parameters. ORFs already accounted for (Table 1), are excluded. The following criteria for function are used: (i) transcription direction matches the Chargaff difference prediction, (ii) cross-over points between the Chargaff difference lines for the W and S bases are positioned near ORF borders, (iii) Chargaff differences are greatest in the middle of the ORF. Most weight is placed on the first criterion. In general the rejection decisions are confirmed, and the featureless regions of the BX-C remain unexplained in terms of protein-encoding capacity.
However, some ORFs should be reconsidered in that they fulfil all three of the criteria. In this respect, we draw attention to leftward ORFs 36, 37, 39, and 72 in intron 1 of Abd-B. Also of particular note are rightward ORFs 30 and 33 which overlap the region in the centromeric flank (around nt 330,000 to 332,000) where, if transcription occurred, it should be to the right (Fig. 1). In view of the generally compact nature of fruit fly gene organization, it seems likely that, if this region is not part of the BX-C (i.e. it consists of DNA organized in the E. coli mode), then the region should be compact and the ORFs are functional.
A striking, but unexplained, feature of the analysis (criterion (i) in Table 3) is that although transcription directions do not conform to, they are not indifferent to, the Chargaff difference predictions. Of the 47 listed ORFS, only 9 are positive. 38 are negative, with a binomial probability P<0.0001.
Optimum sequence window in Drosophila
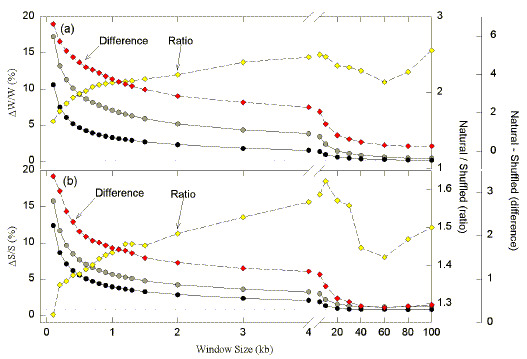
The above analysis employed a 1 kb window size which is optimum when applied to other genomes (e.g. E. coli, S. cerevisiae, vaccinia virus; Forsdyke and Bell, 1997; Bell and Forsdyke, unpublished work). To examine this further, windows sizes were varied while maintaining the 100 nt shift between successive windows. In Fig. 2 average absolute Chargaff differences are plotted against the size of sequence windows. With windows of only 100 nt, high differences would be expected since there would be great statistical fluctuations when base "coins" are "tossed" less than 100 times. Average absolute differences for both the W bases and the S bases are high when windows are 100 nt. Values for the natural sequence exceed those for the corresponding shuffled version of the sequence, implying evolutionary pressures on base order favouring the generation and maintenance of Chargaff differences.
|
|
| Fig. 2. Variation of average absolute Chargaff difference values with size of windows in the 338,234 nt sequence containing the BX-C. Windows of varying size were moved along the sequence in steps of 100 nucleotides, and base compositions were determined in each window. Absolute Chargaff differences were calculated for Fig. 2a as DW/W and expressed as a percentage. DW is the absolute value of the difference between the number of W bases (DW=|A-T|), and W is the sum of the W bases (W=A+T). Absolute Chargaff differences were calculated for Fig. 2b as DS/S and expressed as a percentage. DS is the absolute value of th edifference between the number of S bases (DS=|C-G|), and S is the sum of the S bases (S=C+G). Average Chargaff differences for each window size are plotted either as large grey symbols (natural sequence), or as large black symbols (shuffled sequence). Small open diamonds refer to the ratio of these values (the average Chargaff difference for the natural sequence divided by the average Chargaff difference for the shuffled sequence). Small filled diamonds refer to the difference between these values. The horizontal dotted lines indicate Chargaff differences for the entire sequence (i.e. the largest possible window, of which there is only one copy). Thus, the total number of windows of a given size varies with sequence length. In a 100 kb sequence there will be 999 windows of 0.2 kb, and one window of 100 kb. |
With increasing window size average Chargaff differences for both natural and shuffled sequences decrease exponentially to approach the value for the entire 338,324 nt segment. These are 0.149% for the W bases and 0.861% for the S bases. For the W bases only 1 in 671 bases does not have a potential pairing partner in the same DNA strand. For the S bases 1 in 116 bases does not have a potential pairing partner in the same DNA strand.
The window size at which Chargaff differences for the natural and shuffled sequences diverge maximally should be the most informative with regard to evolutionary selection pressures on the primary sequence of DNA (e.g. the encoding of a protein). Values for the natural and shuffled sequence were compared either as a ratio, or by subtraction. The ratios increased progressively with increasing window size up to 5-10 kb windows, with the curves tending to plateau after 1 kb. The differences were maximum at the lowest window size employed (0.1 kb), and then declined rapidly, tending to plateau at around 1 kb. Although no individual window size emerged as optimum, in the present work window sizes of 1 kb were chosen as a working compromise to maintain consistency with our studies in other species where the optimum window is more apparent. Indeed, Chargaff difference plots for fruit fly sequences using window sizes lower (e.g. 0.5 kb) and higher (e.g. 2 kb) than 1 kb, were found to be less informative (data not shown).
Discussion
![]()
While the compact "E. coli mode" of genome organization is maintained in the flanks of the BX-C, the complex itself seems more in the "mammalian mode", with long stretches of intergenic DNA and long introns. It may be feasible to dismiss mammalian mode DNA in mammals as "junk" (since it is characteristic of the entire genome), but this is not so easy in the case of mammalian mode DNA in the compact fruit fly genome.Thus, there must be something special about the BX-C, which has allowed it to escape the genome-wide evolutionary pressure for the elimination of DNA segments which appear to be of little or no importance for reproductive success (Petrov et al. 1996). Understanding the organization of mammalian mode DNA in the fruit fly genome may allow a better understanding of mammalian mode DNA in mammals.
The Chargaff difference transcription rule applies to various compact genomes of organisms such as E. coli, S. cerevisiae, SV40 virus, and vaccinia virus (Szybalski et al. 1966; Rudner et al. 1969; Forsdyke and Bell, 1997; Bell and Forsdyke, 1999a,b). That the rule might also apply to apparently less compact mammalian genomes or genome segments is evident from previous studies of various mammalian genes (Smithies et al. 1981; Mrazek and Kypr 1994), and from our unpublished studies of the human "G0 /G1 Switch 2" gene (G0S2; Russell and Forsdyke 1991; Cristillo et al. 1997). In this light, it would be expected that most of the genes under study, both in the BX-C and in its flanks, would obey the transcription rule, as is indeed found. Thus, the fruit fly evidence (Tables 1, 2) further supports the general validity of the Chargaff difference transcription rule. However, as in the other species studied, a minority of genes appear to violate the rule.
A notable example is an intronless gene in the BX-C (glut-l), predicted by Martin et al. (1995) to correspond to a human gene encoding a glucose transporter type 3 (GLUT3; 23.9% identity). This assignment is not supported by Chargaff difference analysis in terms of transcription direction (Table 1), but is strongly supported by large Chargaff difference values near the postulated centre, and by cross-over points near possible transcription borders (data not shown).
| It should be noted that, although the leftward transcribing glut-l ORF is the longest in the region, there is an overlapping ORF (number 17 in Table 3) of 190 amino acids which is transcribed to the right. If this were the primary transcript there would be no conflict. Another possible explanation would be that the glut-l gene is transcribed to the left to generate an "antisense" transcript with T-rich loops. The apparent GLUT3 homolog might function as an "antisense" RNA which might inhibit glucose transporter mRNAs with A-rich loops transcribed from other fruit fly genes; (the reason for the protein homology might be that sometimes sense and antisense strands encode similar peptide segments; Forsdyke 1995). One of the two BX-C genes considered to function at the RNA level, the leftward transcribing iab-4 gene, also violates the transcription rule, again suggesting a possible antisense role. |
The two modes of genome organization (E. coli and mammalian) in the BXC segment under study are supported by Chargaff difference analysis of the unassigned ORFs recognized by Martin et al. (1995). An apparent lack of compactness in the centromeric flank (Fig. 1) may be illusory, since ORFs 30 and 33 (around nt 330,000-332,000; Table 3), which were discarded by Martin et al. (1995), conform well to Chargaff difference criteria for function (transcription direction, cross-over points near possible transcription borders, large values near the ORF centre,). In view of this conformity it is surprising that they are not a single ORF, rather than two independent ORFs.
| The lack of compactness of the BX-C, cannot be easily explained in terms of unassigned ORFs (Table 3). The pattern of curves shown in Fig. 1 for a region mainly consisting of the centromeric flank (E. coli mode DNA), is, in its general appearance, quite similar to patterns observed in the BX-C in intergenic DNA and introns (data not shown). Since the patterns in the flanks correlate with regions of known function, it is possible that the patterns we observe in unassigned regions of the complex are providing information on other functions (some of which might require transcription, but not translation). Some of these functions would be regulatory (Lewis et al. 1995). |
Hybridization studies suggest that the member of a Watson-Crick base pair for which a strand is enriched, is clustered (Szybalski et al. 1966). This might to some extent account for the many W-rich clusters observed in the BT-X (Lewis et al. 1995). The clusters are presumed to contribute, partly or entirely, to the deviations from Chargaff's second parity rule described here. When DNA is subjected to extensive negative supercoiling there is the possibility of extrusion of single-standed stem-loop structures (Murchie et al. 1992; Forsdyke, 1996; Kleckner, 1997). Since, by definition, the base composition of stems would closely follow parity, it is likely that deviations from parity correspond to the loop regions of potential stem-loop structures (Fig. 3). Indeed, our DNA folding studies (Zuker, 1994) show that Chargaff differences closely correlate with loop compositions (r = 0.98; Bell and Forsdyke, 1999a,b).
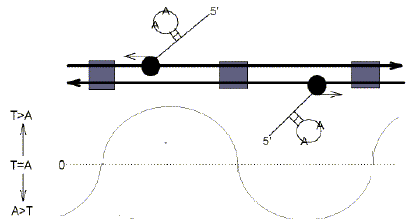 |
| Fig. 3. In Drosophila melanogaster the direction of transcription should correlate with the relative A- or T-richness of the loops in potential stem-loop structures which might be formed by the "top" DNA strand. Duplex DNA (long parallel left- and right-pointing arrows representing the upper and lower strands and their 5'->3' polarities) is transcribed by RNA polymerases (black balls with trailing nascent RNAs), which may utilize as template either upper or lower strands (small arrows indicating direction of transcription), and will transcribe RNA with the potential for A-rich loops only. Grey boxes refer to non-transcribed DNA containing regulatory elements (e.g. promoters). The curve fluctuating above and below the zero line indicates Chargaff differences for the top DNA strand. |
Whether a postulated ORF is actually functional is best decided by the direct identification of a functional gene product. A decision to embark on this laborious process is easier if the ORF accords to indirect criteria such as codon choice and minimum length. Chargaff difference analysis merely requires determination of base composition, and provides a simple additional criterion for ORF identification.
Acknowledgements
We thank S. Celniker for providing her list of putative ORFs, L. Russell for technical help, A. Cristillo and J. Gerlach for assistance with computer configuration, T. Smith for statistical advice, and P. Sibbold for review of the manuscript. The work was supported by grants from the Medical Reseach Council of Canada, and Queen's University.
References
Chargaff, E. 1951. Structure and function of nucleic acids as cell constituents. Fed. Proc. 10: 654-659.
Chargaff, E. 1979. How genetics got a chemical education. Ann. N. Y. Acad. Sci. 325: 345-360.
Cristillo, A. D., Heximer, S. P., Russell, L., and Forsdyke, D. R. 1997. Cyclosporin A Inhibits Early mRNA Expression of G0 /G1 Switch Gene 2 (G0S2) in Cultured Human Blood Mononuclear Cells. DNA Cell. Biol. 16, 1449-1458.
Forsdyke, D. R. 1995. Sense in antisense? J. Mol. Evol. 41: 582-586.
Forsdyke, D. R. 1996. Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths". J. Theor. Biol. 178: 405-417.
Forsdyke, D. R., and Bell, S. J. 1997. Deviations from Chargaff's second rule correlate with direction of transcription and genome structure. Proc. Can. Fed. Biol. Socs. 40: 87 (abstract no. 260).
Gribskov, M., and Devereux, J. 1991. Sequence Analysis Primer. Stockton, New York.
Karkas, J. D., Rudner, R., and Chargaff, E. 1968. Separation of B. subtilis DNA into complementary strands. II. Template functions and composition as determined by transcription by RNA polymerase. Proc. Natl. Acad. Sci. USA 60: 915-920.
Karlin, S., and Mrazek, J. 1996. What drives codon choices in human genes? J. Mol. Biol. 262: 459-471.
Kleckner, N. 1997. Interactions between and along chromosomes during meiosis. Harvey Lectures 91: 21-45.
Lewis, E. B. 1992. Clusters of master control genes regulate the development of higher organisms. J. Amer. Med. Assoc. 267: 1524-1531.
Lewis, E. B., Knafels, J. D., Mathog, D. R., and Celniker, S. E. 1995. Sequence analysis of the cis-regulatory regions of the bithorax complex of Drosophila. Proc. Natl. Acad. Sci. USA 92: 8403-8407.
Martin, C. H., Mayeda, C. A., Davis, C. A., Ericsson, C. L., Knafels, J. D., Mathog, D. R., Celniker, S. E., Lewis, E. B., and Palazzolo, M. J. 1995. Complete sequence of the bithorax complex of Drosophila. Proc. Natl. Acad. Sci. USA 92: 8398-8402.
Meyer, R. K., and Krueger, D. D. 1994. Minitab Computer Suppliment. Macmillan College Publishing Co, New York.
Mrazek J., and Kypr, J. 1994. Biased distribution of adenine and thymine in gene nucleotide sequences. J. Mol. Evol. 39: 439-447.
Murchie, A. I. H., Bowater, R., Aboul-Ela, F., and Lilley, D. M. J. 1992. Helix opening transitions in supercoiled DNA. Biochem. Biophys. Acta 1131: 1-15.
Oliver, S. G. et al. 1992. The complete DNA sequence of yeast chromosome III. Nature 357: 38-46.
Petrov, D. A., Lozovskaya, E. R., and Hartl, D. L. 1996. High intrinsic rate of DNA loss in Drosophila. Nature 384: 346-349.
Prabhu, V. V. 1993. Symmetry observations in long nucleotide sequences. Nucleic Acids Res. 21: 2797-2800.
Rudner, R., Karkas, J. D., and Chargaff, E. 1969. Separation of microbial deoxyribonucleic acids into complementary strands. Proc. Natl. Acad. Sci. USA 63: 152-159.
Russell, L., and Forsdyke, D. R. 1991. A human putative lymphocyte G0 /G1 switch gene containing a CpG-rich island encodes a small basis protein with the potential to be phosphorylated. DNA Cell Biol. 10: 581-591.
Smithies, O., Engels, W. R., Devereux, J. R., Slightom, J. L., and Shen, S. 1981. Base substitutions, length differences, and DNA strand asymmetries in the human Gl and Al fetal globin gene region. Cell 26: 345-353.
Szybalski, W., Kubinski, H., and Sheldrick, P. 1966. Pyrimidine clusters on the transcribing strands of DNA and their possible role in the initiation of RNA synthesis. Cold Spring Harbor Symp. Quant. Biol. 31: 123-127.
Watson, J. D., and Crick, F. H. C. 1953. Genetical implications of the structure of deoxribonucleic acid. Nature 171: 964-967.
Zuker, M. 1994. Prediction of RNA secondary structure by energy minimization. In Computer Analysis of Sequence Data, Part II. Ed. Griffin, A. M., and Griffin, H. G. pp. 267-294. CRC Press, Totowa, NJ.
![]()
Return to: HomePage (Click Here)
Return to: Bioinformatics Page (Click Here)
![]()
Posted circa 1999 and last edited 29 November 2005 by D. R. Forsdyke
![]()