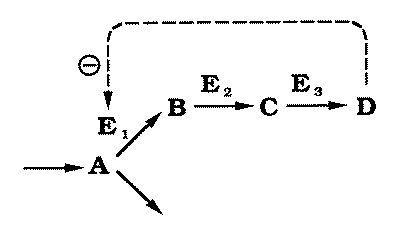
FIG. 2. A hypothetical
metabolic pathway catalyzed by enzymes E1, E2 and E3.
A is the substrate of E1,
which catalyzes the rate-limiting step in the pathway. A is also metabolized by
another pathway. B is the product of E1
and the substrate of E2, which is
normally not rate limiting. C is the product of E2 and the substrate of E3,
which is also normally not rate limiting. D, the product of E3, is responsible for the phenotype and
for pathway regulation by feedback inhibiting E1. |
Substrate A is converted to end-product D through a
series of intermediates (B, C), catalyzed by enzymes E1,
E2, and E3. The first step of the pathway, the rate-limiting step,
is subject to feedback inhibition by D. Using radioactively labelled A
and short incubation periods, it has been shown experimentally that the system may be
modeled as if in vivo the intermediate steps had no effect on the reaction rate
(Forsdyke, 1971). |
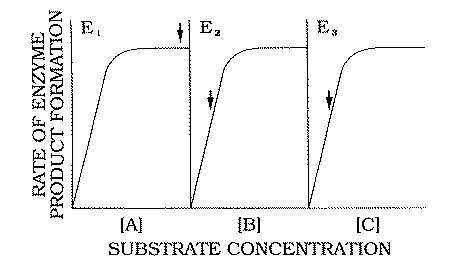
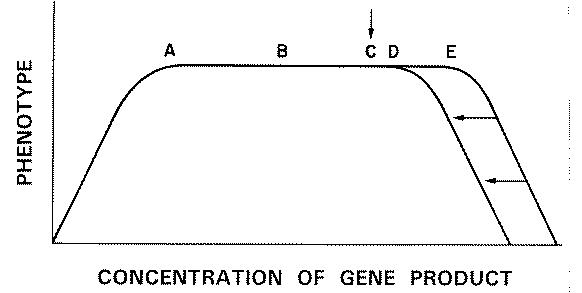
FIG. 3. Hypothetical in
vivo dose-response curves for enzymes E1 , E2 , and E3
. Rates of formation of products (B, C, and D) are expressed as
functions of the concentrations of substrates A, B, and C,
respectively. The vertical arrows refer to the normal in vivo concentrations of these
substrates as they interact with the enzymes. |
|
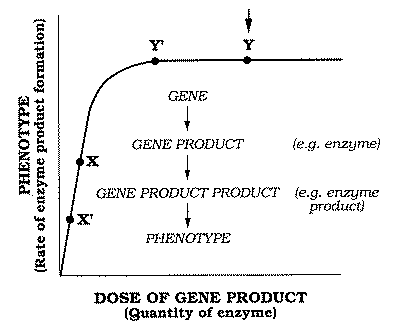
Figure 3 shows hypothetical in vivo substrate dose-response curves
for the three steps in the pathway. The vertical arrows indicate the normal substrate
concentration existing in vivo. In the case of the rate-limiting enzyme El
the in vivo concentration of A must, by definition, correspond with the
plateau of the dose-response curve, so that enzyme concentration, and not substrate
concentration, is rate limiting. This would correspond to point X on the
plot of reaction rate versus enzyme concentration (Fig. 1).
In the case of the non-rate-limiting
enzymes E2 and E3, the normal substrate concentration corresponds to
the ascending limbs of the corresponding substrate dose-response curves (Fig. 3). There is
ample enzyme to accommodate fluctuations in availability of the substrates B
and C (the products of El and E2, respectively).
This quantity of enzyme would correspond to point Y on a plot of reaction
rate versus enzyme concentration (Fig. 1).
Thus, after a molecule of A
has squeezed through the "bottle-neck" El
to become B, subsequent chemical modifications by E2 and E3
do not influence the rate of accumulation of the end-product D.
It can be seen that the situation with the non-rate-limiting enzymes (E2,
E3) corresponds to the "margin-of-safety"
scenario. In the case of the rate-limiting enzyme E1, halving of gene-product
concentration (X to X' in Fig. 1) would have
consequences for the phenotype.
However, in general, rate-limiting enzymes are subject to
complex controls by products of intermediary metabolism. The most common of these is
end-product inhibition (Fig. 2). A decrease in D would decrease the
inhibition. This would increase the activity per molecule of E1, so that
the reaction rate would become the same as in the homozygote. Thus, in this case, feedback
inhibition provides another "margin of safety"
allowing a heterozygote to maintain the wild-type phenotype.
|
4.
"Extreme Environmental Disturbances"
A major problem with what we might now call the margin-of-safety theory, is in determining what selective
forces would have created and sustained the margins of safety. Some have argued that this
can be explained entirely in metabolic terms (Kacser & Burns,
1980). In the case of a
rate-limiting enzyme (E1) the margin could indeed be a simple consequence of the evolution
of metabolic controls. It is in the case of non-rate-limiting enzymes that a further
explanation must be sought. What sustains the enzyme activity in a wild-type homozygote at
point Y rather than at point Y' (Fig. 1)?
Haldane interpreted dominance in metabolic terms, but adopted
Fisher's view that weak selective evolutionary forces acting on heterozygotes would be sufficient to maintain the
margin of safety. Thus Haldane (1930) wrote: |
 |
"If we imagine a race whose genes were only just doing the
work required of them, then any inactivation of one of a pair of genes would lead to a
loss of total activity. Thus if "A1
A 1"
can just oxidize all of a certain substrate as fast as it is formed, its inactivation will
produce a zygote "A1 a"
which can only oxidize about half. If now A1
mutates to A2, which can oxidize
at twice or thrice the rate of A1,
if necessary, no effect will be produced, i.e. "A1 A2"
and "A2 A2" zygotes
will be indistinguishable from "A1
A1".
But "A2 a" will be
normal. Hence "A2 a"
zygotes will have a better chance of survival than "A1 a", and
A2
will be selected."
|
 |
Muller (1932), however, thought that the evolutionary selection would act
at the homozygote level. He
postulated:
"that the mutations favouring dominance .. . have been
selected and are maintained, not so much for their specific protection against
heterozygosis at the locus in question, but as to provide a margin of stability
and security, to insure the organism against weakening or excessive
variability of the character by other and more common influences -- environic and probably
also genetic".
|
|
Along similar lines Wright (1977) stated that:
"because of extreme environmental disturbances,
a considerable excess [of gene product] is advantageous. This is likely to be so great
that the correlated response of the rare heterozygote is also brought fairly close to the
asymptote, thus giving a high degree of dominance."
The possible nature of the "extreme environmental
disturbances" was not specified.
|
 |
5. The Heat-shock
Response
|
The heat-shock response follows a sudden change in various physical
or chemical features of the environment, and is particularly notable following an increase
in temperature (Nover, 1989). The response is detected as a rapid increase in the
intracellular concentrations of a set of evolutionarily conserved "heat-shock" proteins, which is accompanied by a decrease in the concentrations of most normal proteins. The
notion that the response is predominantly concerned
with protection against thermal and other types of "stress"
has recently lost ground (Bader et al., 1992; Fisher et al., 1992).
It is proposed elsewhere (Forsdyke, 1985, 1991, 1992,
1995), that
the response has evolved as part of a mechanism for distinguishing the proteins of
intracellular pathogens ("not-self")
from normal intracellular proteins ("self").
The proteins of the crowded cytosol exert a collective pressure tending to make individual
protein species aggregate when their concentrations exceed their individual solubility
limits. These concentrations have been fine-tuned over evolutionary time so as not to
exceed these limits. Not-self proteins more readily "trip"
an intracellular surveillance system because their concentrations have not been so fine
tuned.
The aggregations, however, being primarily entropy driven
(Lauffer, 1975), are strongly increased by an increase in temperature. The organism exploits this
(e.g. fever) to promote the aggregation of the proteins of a foreign pathogen (Nguyen et
al., 1989). In this process self proteins might also be aggregated. To avoid this, the
concentrations of normal proteins decrease. However, in turn, this decreases the
collective pressure exerted by the cytosolic proteins to make the proteins of the pathogen
aggregate. To compensate for this, a special set of proteins, the heat-shock proteins, are
produced (Forsdyke, 1995).
The main point to be made about the heat-shock response in the
present context is that it probably reflects a fundamental process which appeared early in
evolution when sets of replicators encased in a membrane (prototypic cells) had to be
protected against invasion by foreign replicators (prototypic viruses; Forsdyke,
1991).
All subsequent evolutionary developments would potentially be influenced by this
pre-existing system. The sudden general fall in the concentration of normal self proteins
as part of the heat-shock response would severely compromise cell function if there were
not a margin of safety regarding function. Thus the heat-shock response would constitute a
powerful evolutionary force acting on wild-type homozygotes.
This would lead to the
general evolution of proteins of a specific activity sufficient to sustain (or facilitate
the recovery of), cell function, at a time when protein concentration had fallen. An
incidental outcome of this would be that a heterozygote would normally have sufficient
gene product so that it would be phenotypically indistinguishable from the wild type.
However, having only one copy of the allele in question, a
heterozygote would have sacrificed part of its margin of safety and this might have some
impact on viability. Indeed, heterozygotes for lethal mutations do show some reduction in
viability compared with the homozygous wild types and may be more temperature-sensitive
(Plunkett, 1932; Simmons & Crow,
1977).
A viral infection might trigger a fever and an
associated heat-shock response; the quantity of a non-rate-limiting protein might then
fall from level Y' to X in Fig. 1. Thus a protein unable
to respond to normal metabolic controls (e.g. show increased activity in response to a
loss of end-product inhibition), would now become rate limiting. The consequence of this
would depend on timing and the nature of the end-product involved. A viral infection at a
key developmental stage might have disastrous consequences.
|
6.
X-Chromosome Dosage Compensation
The hypothesis offers a new way of looking at the problem
of X-chromosome dosage compensation. This was first described as the process by which the
function of the single X-chromosome in male fruit flies is made equivalent to the function
of both X-chromosomes in females (Muller,
1948). Without dosage compensation, the
situation would be formally equivalent to the points Y (in females) and Y'
(in males) as shown in Fig. 1.
Muller postulated that these "exceedingly minute" phenotypic differences
(differences between the point Y and point Y'
phenotypes) would constitute a sufficient selection pressure for dosage compensation to
have evolved. The heat-shock response, however, in shifting heterozygote (male) gene
product concentrations from point Y' to point X (Fig.
1), would have created a much greater selection force for the evolution of a margin of
safety in males. Although this might have been a factor in the evolution of dosage
compensation, it is argued in the accompanying paper (Forsdyke,
1994) that the major
factor is probably the need to fine-tune protein concentrations,
rather than protein functions, to be equal in male and female cells. |
This work was supported by the Medical Research Council of
Canada and the Leukaemia Research Fund of Toronto.
REFERENCES
BADER, S. B., PRICE, B. D., MANNHEIM-RODMAN, L. &
CALDERWOOD, S. K. (1992). Inhibition of heat-shock gene expression does not block the
development of thermotolerance. J. Cell. Physiol.
151, 56-62.
CHARLESWORTH, B. (1979). Evidence against Fisher's theory of
dominance. Nature, Lond. 278, 848-849.
FISHER, B., KRAFT, P., HAHN, G. M. & ANDERSON, R. L. (1992).
Thermotolerance in the absence of induced heat-shock proteins in a murine lymphoma. Cancer Res. 52, 2854-2861.
FISHER, R. A. (1931). The evolution of dominance. Biol. Rev. 6, 345-368.
FORSDYKE, D. R. (1971). Application of the isotope-dilution
principle to the analysis of factors affecting the incorporation of 3H-uridine
and 3H-cytidine into cultured lymphocytes.
Biochem. J. 125, 721-732.
FORSDYKE, D. R. (1985). Heat-shock proteins defend against
intracellular pathogens: a non-immunological basis for self/not-self discrimination. J.
theor. Biol. 115, 471-473.
FORSDYKE, D. R. (1991). Early evolution of MHC polymorphism. J. theor. Biol. 150, 451-456 (1991).
FORSDYKE, D. R. (1992).Two signal model of self/not-self
discrimination: an update. J. theor. Biol.
154, 109-118.
FORSDYKE, D. R. (1994). Relationship of X chromosome dosage
compensation to intracellular self/not-self discrimination: a resolution of Muller's
paradox. J. theor. Biol. 167,
7-12.
FORSDYKE, D. R. (1995). Entropy-driven protein self aggregation
as the basis for self/not-self discrimination in the crowded cytosol. J. Biol. Systems 3, 273-287.
HALDANE, J. B. S. (1930). A note on Fisher's theory of the
origin of dominance and on a correlation between dominance and linkage. Am. Nat. 64, 87-90.
KACSER, H. & BURNS, J. A. (1980). The molecular basis of
dominance. Genetics 97, 639
666.
LAUFFER, M. A. (1975). Entropy-driven
Processes in Biology. New York: Springer-Verlag.
MENDEL, G. (1866). Versuche uber Pfanzenhybriden. Verhandl. Naturforsch. Ver. Brunn 4,
3-47 (1866).
MULLER, H. J. (1932). Further studies on the nature and causes
of gene mutations. In: Proc. 6th Int. Cong. Genet.
2, (Jones, D. F. ed.) pp. 213-255. Menasha, WI: Banta.
MULLER, H. J, (1948). Evidence on the precision of genetic
adaptation. Harvey Lect. 43, 165-229.
NGUYEN, V. T., MORANGE, M. & BENSAUDE, 0. (1989). Protein
denaturation during heat shock and related stress. E. coli beta-galactosidase and Photinus
puralis luciferase inactivation in mouse cells. J.
Biol. Chem. 264, 10487-10492.
NOVER, L. (1989). 125 years of experimental heat-shock research.
Genome 31, 668-670.
ORR, H. A. (I 99 1). A test of Fisher's theory of dominance. Proc. natn. Acad. Sci. U.S.A. 88,
11413-11415.
PLUNKETT, C. R. (1932). Temperature as a tool of research in
phenogenetics. In: Proc. 6th Int. Cong. Genet.
2, (Jones, D. F. ed.), pp. 158-160. Menasha, WI: Banta.
SIMMONS, M. J. & CROW, J. F. (1977). Mutations affecting
fitness in Drosophila populations. A. Rev. Genet.
11, 49-78.
WRIGHT, S. (1977). The evolution of dominance. In: Evolution and the Genetics of populations, Vol,
3, pp. 498-526. Chicago: The University of Chicago Press.
| End Note
The
basic idea in this paper was advanced by Hugo de Vries (see Treasure
Your Exceptions;
Chapter 13)
and in 1909 by
George H. Shull (The
"presence and absence" hypothesis. American
Naturalist 43,
410-419).
| "Having arrived at the
conclusion that all the Mendelian characters are dependent upon
chemical relations, we may return to the question of dominance,
and the relation between the two kinds of homozygotes and the
heterozygote, and see to what extent the known facts may be
interpreted in terms of chemical experience.
A fundamental principle in this connection
is the law that the extent of a reaction between two chemicals
is determined by the amount of the reagent which is present in
less relative quantity, and not by the one which is present in
excess. When the positive homozygote ... and the heterozygote
... are alike, i.e., when there is complete dominance of
presence over absence, it may mean that already the presence of
the one unit A of the heterozygote is sufficient to result in
the maximum reaction, in which case the doubled factor AA of the
positive homozygote can do no more.
When, on the other hand, one unit A is not
sufficient to produce a maximum reaction with the other factors
present, the AA of the homozygote produces the corresponding
character in greater intensity, and the heterozygote will be
intermediate between the two homozygous parents." |
Regarding his dispute with Wright, in correspondence (1934) Fisher
wrote:
"It is quite obvious that in a chemical reaction one ingredient
may be present in excess, in the sense that small variations in its
amount have very little effect on the speed of the reaction, while a
large diminution of it would slow the reaction down. That this is
probably the case with the products of some genes is shown by Stern's
'bobbed' allelomorphs.
It is a relatively obvious way of producing
dominance against mutations which partially inactivate the mutant genes.
It might, as far as my theory is concerned, be the only mechanism by
which dominance is produced, though I do not imagine that this is so.
But if this were so, the occurrence of dominance would be just as much
in need of explanation as if dominance were produced by some other
mechanism. For the fact that one component of a reaction is present in
excess implies that its speed is regulated by other components, and that
mutations affecting these, if they occurred, would not be recessive,
whether the mutation reduced the activity or enhanced it.
On the theory
of components in excess we should have to say that the organism had been
so modified that the speeds of all biochemical processes were regulated
only by the products of genes incapable of mutation."
Natural Selection, Heredity and Eugenics. Edited by J. H.
Bennett. Oxford Univ. Press 1983. p. 235 |
2. Dosage compensation
Relationship of
X-Chromosome Dosage Compensation to Intracellular Self/Not-self Discrimination: A
Resolution of Muller's Paradox?
The Journal of Theoretical Biology (1994) 167,
7-12.
(With copyright permission from Academic Press)
(Received on 10 April 1992, accepted in revised form on 3 June 1993)
1. Introduction
2. Aneuploidy and the
Evolution of Dosage Compensation
3. "Exceedingly Minute
Differences"
4. Fine-tuning of
Intracellular Protein Concentrations
5. Differential Aggregation of
Foreign Proteins
6. Role of X Chromosome Inactivation
7. Two Active X Chromosomes
in Oocytes and Embryos
8. Compensation in Z/W Chromosome
Systems
ADDED NOTE: 2001
ADDED NOTE: 2005
ADDED NOTE: May 2008
ADDED NOTE: Sept 2008
ADDED NOTE: Nov 2008
Abstract.
Muller's paradox is that X chromosome dosage compensation seems to have evolved in spite
of there being only "exceedingly minute differences"
between compensated and uncompensated phenotypes. The paradox can be resolved by
considering, not the specific functions of individual proteins, but the collective functions of proteins per se.
One
such function could be the collective pressure exerted by proteins in the crowded cytosol
to drive individual protein species from simple solution (aggregation) when their
concentrations exceed specific thresholds. It is proposed that over evolutionary time,
individual genes, both on X-chromosomes and autosomes, would have fine-tuned factors such
as transcription rates and protein stabilities to this collective pressure.
However,
without X-chromosome dosage compensation the total concentration of cytosolic proteins,
and hence the collective pressure, would have fluctuated between male and female
generations. Fine-tuning, a process of vital importance for intracellular self/not-self
discrimination, would have been severely compromised.
|
|
1. Introduction
In many species the sex of an individual depends on
which of two alternative sex chromosomes he/she inherits ( Bull,
1983). In some species
(e.g. fish, amphibia) the sex chromosomes are similar (homomorphic) and appear to differ
only in the genes affecting sexual differentiation. The latter are kept from recombining
with their alleles by inversions or by some other mechanism (Ohno,
1967). In other species
(e.g. mammals, birds) the sex chromosomes differ in size (heteromorphic). One of the
chromosomes appears to have degenerated losing most of its genes (Charlesworth,
1991). One
sex, the homogametic sex, contains two copies of the normal, undegenerate, chromosome (X
or Z). The other sex, the heterogametic sex, contains one normal chromosome and one
degenerate chromosome (Y or W). These show sequence similarity only in a short (pseudoautosomal) segment.
The essential haploidy
(hemizygosity) of the sex
chromosomes in the heterogametic sex has three important implications:
(i) Deleterious recessive mutations are expressed. Individuals
containing these mutations will tend not to reproduce and so fewer deleterious mutations
should be passed on to future generations.
(ii) It is not possible to repair DNA damage (or excise transposed
elements; Steinemann, 1982) on the basis of information contained in an allelic
chromosome. Without accurate repair the load of deleterious mutations passed on to future
generations would increase (Bernstein & Bernstein,
1990). Eternally locked in
heterogametic generations, the Y and W chromosomes would never have had the opportunity to
repair damage on this basis and this may have contributed to their degeneration.
(iii) Since gene dosage is often a major determinant of gene product
dosage, the dosage of X- and Z-encoded gene products (proteins) in the heterogametic sex
should be half those in the homogametic sex. This discrepancy might either be accepted by
the organism (dosage tolerance or dosage imbalance), or be adjusted (dosage compensation
or dosage balance).
Studies by Muller with the fruit fly Drosophila
melanogaster in the late 1920s established the existence of a mechanism to adjust
the concentrations of X-encoded gene products (excluding some concerned with sexual
differentiation and a few others), so that these were equalized between the sexes
( Muller, 1948).
It is now known that this involves an increase in the expression of the single
X-chromosome in the male fruit fly to equal the combined expression of the two X
chromosomes in the female (Kuroda et al., 1991). In mammals equalization is
achieved by inactivating one of the X chromosomes in the female (Lyon,
1992).
A necessary condition for the evolution of dosage
compensation was the degeneration of one of the sex chromosomes to create the
Y-chromosome. However, with no clear justification, the assumption has grown that the
degeneration of the Y-chromosome was alone sufficient
to drive the evolution of dosage compensation. Thus Haldane ( 1933) wrote:
"The tendency to balance may be regarded as a secondary
effect of the accumulation of recessive lethals on the Y-chromosome. Indeed, this
accumulation can only proceed as the X develops internal balance"
(my italics).
This coupling of Y degeneration and dosage compensation is implied by some modern
authors. Charlesworth ( 1978) wrote that the gradual degeneration of the Drosophila Y-chromosome
itself:
"Creates a selection pressure for
differentially increasing the activity of the X-chromosome in heterogametic individuals"
(my italics).
Of the mammalian type of dosage compensation, he wrote
( Charlesworth, 1991) that this:
" Creates a selective advantage to
reducing X activity in the homogametic sex" (my
italics),
and added that
degeneration of the Y-chromosome and dosage compensation are "
processes
which must almost inevitably occur"
This paper describes the attempts of Muller and others to
understand the forces driving the evolution of dosage
compensation. It is argued that dosage compensation can best be understood by taking into
account the possibility that proteins influence phenotypes not only on the basis of their
individual specific functions, but
also on the basis of their collective
functions as proteins per se. While still in need of experimental verification, the
strength of this paper's hypothesis is that it brings together a variety of observations
in fields which have not previously been seen as related.
2.
Aneuploidy and the Evolution of Dosage Compensation
The issue of what drove the evolution of dosage
compensation was partially addressed by Ohno ( 1967) who noted that a heterogametic
individual can be regarded as aneuploid for the X-chromosome.
"The hemizygous existence for all the genes on the X should
be very perilous since monosomy for even the smallest autosome (only one fourth the size
of the X) is apparently lethal in man".
Why aneuploid states are lethal was not discussed. In Drosophila Lindsley and
coworkers ( 1972) found that:
"the deleterious effects of aneuploidy are, in the main,
caused by the additive effects
of genes that slightly reduce
viability and not by the individual effects of a few
aneuploid-lethal genes among a large array of dosage-insensitive loci"
( my italics).
Orr ( 1990), when considering why polyploidy is rarer in animals than in
plants, took the issue further. He noted first:
"The difficulty in establishing a tetraploid line in
organisms with a genetically degenerate sex chromosome: although polyploid speciation does
not necessarily disrupt sex determination in such species, it does
invariably disrupt the balance of X chromosome relative to autosomal gene products
normally maintained by dosage compensation" ( Orr's
italics).
He noted further that in mammalian females:
"The number of Xs that remain active increases with the
number of autosomal sets"
He then concluded that:
"The doses of X-linked and autosomal genes have been
fine-tuned
by natural selection to ensure proper interactions
between
these loci" (my italics).
The nature of the "proper interactions" and why
they had to be "fine-tuned" were not discussed.
The importance of the ratio of sex chromosomes to autosomes both for dosage compensation and for sex-determination
is well recognized in fruit fly and nematode systems ( Hodgkin,
1990). Since in mammalian
systems ratio-dependence seems to have been retained only for dosage compensation
(Book
& Santesson, 1961; Harnden, 1961; Jacobs & Midgeon, 1989), it is likely to be
critical for this process.
It is difficult to imagine how sex chromosomes and autosomes
could sense each other's dosage directly, and it seems more likely that the quantitation
is carried out at the gene-product level. Furthermore, noting that polyploid cells are
larger than euploid cells (thus presumably keeping the concentration of cytoplasmic
components constant), it seems likely that the quantitation involves some relationship
between the concentrations of the products of sex chromosomes and autosomes (rather than a
relationship between their absolute quantities).
3. "Exceedingly Minute Differences"
Muller ( 1948) approached the problem of the
evolution of dosage compensation in fruit flies by first noting the relationship between
the concentration of a protein and the activity of that protein as observed in a
quantifiable phenotype. As a dose of a gene (and hence of the corresponding protein) is
increased, there would be an approximately linear increase in the character assayed.
However, this would only hold as long as the dose of the protein was limiting. Above a
certain dose something else would become limiting, and the dose-response curve would tend
to plateau (see Forsdyke, 1994 for further
elaboration). Muller noted that the dose of
most wild-type gene products corresponds to a point well along the plateau, so that a
decrease in dose by half, due to hemizygosity, would still leave sufficient gene product
to guarantee maximum activity (i.e. the phenotype would still correspond to a point on the
plateau of the dose-response curve). In this circumstance there would have been no
selection pressure, based on differential gene-product function, for dosage compensation
to have evolved. He described this paradox as follows:
"The effects of individual genes, whether on the X or other
chromosomes, are so near their saturation levels as to make direct discrimination between
one and two doses impossible. Should not the very fact that most of these
genes are so near their saturation level make dosage compensation unnecessary? Why
should there be a perceptible advantage in going through the motions of equalizing them
still further?"
The paradox was not formally acknowledged by Muller
because the following quasi-metaphysical answer, understandable at a time when little was
known of the inner workings of the cell, appeared to satisfy not only Muller, but also his
critics:
"The compensation mechanism must be concerned with the
equalization of exceedingly minute differences. Dosage compensation has in fact become
established because of its advantage in regulating more precisely the grade of characters
whose variations in grade, even without it,
would be exceedingly
minute" (Muller's italics). "The selective forces that established it must depend on such
minute advantages".
No less paradoxical were subsequent studies in other
species (Ohno, 1967). Some X-linked genes encode enzymes. While several enzymes may
contribute to a metabolic pathway, only one of these is normally rate-limiting (Forsdyke,
1994). This rate-limiting enzyme is subject to numerous fine controls, such as end-product
inhibition, resulting in large variations in activity (e.g. 10-fold). Even if an X-linked
enzyme were normally rate limiting, it is unlikely that a two-fold change in enzyme
concentration would affect flow along the metabolic pathway.
Indeed, it is a common
observation that heterozygotic carriers of various traits are often no worse off than the
corresponding homozygotes (Ohno, 1973). Most deleterious mutations show a high degree of
recessivity. Thus, on the basis of these studies also, phenotypic differences upon which
the selective forces of evolution could have acted are unlikely to have been significant.
Along these lines, Chandra (1985) has argued that X-chromosome inactivation is primarily a
sex-determining device and that any compensating effects are incidental.
4. Fine-tuning of Intracellular Protein Concentrations
A consideration of the fine-tuning of the
concentrations of individual proteins within cells, discussed fully elsewhere (Forsdyke,
1995; click here),
provides a possible solution to Muller's paradox. Over a series of generations, a
Y-chromosome and its descendants exist in a succession of male cells. This path may be
expressed as:
Mà Mà
Mà Mà Mà MàMà
Mà Mà Mà Mà Mà
Mà M..…
Over evolutionary time, factors such as the transcription rates of genes on the
Y-chromosome and the stabilities of their products (mRNAs and proteins) could have become
fine-tuned to the needs of this relatively stable intracellular environment. A relatively
constant constitutive concentration of each gene product could have become established. On
the other hand, the X-chromosomes and the autosomes and their descendants alternate
between male and female cells. A typical path might be:
Mà Fà
Mà Fà Fà MàFà
Mà Fà Mà
Fà Fà
Mà M..…
Over evolutionary time it would be difficult to fine-tune both for cells containing a
second X-chromosome and for cells containing a solitary X-chromosome. By inactivating one
X whenever two are present (the mammalian model), or hyperactivating the X whenever one is
present (the fruit fly model), the intracellular environment would be stabilized and
fine-tuning could occur.
|
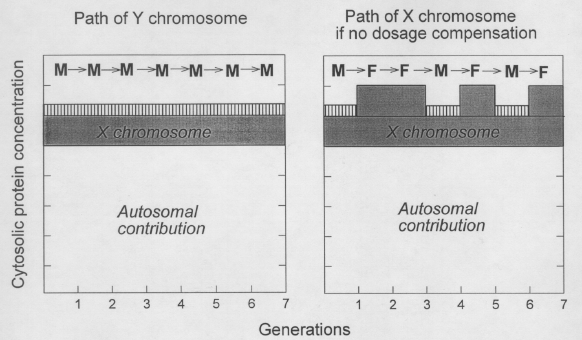
FIG. 1.
Fine tuning of
protein concentration is not possible without X-chromosome dosage compensation.
Passage of
Y-chromosomes and X-chromosomes through the generations occurs either in male (M) or
female (F) cells. Contributions of chromosomes to the cytosolic protein concentration are
shown for autosomes, for X-chromosomes (grey), and for Y-chromosomes (vertical
stripes).
The contribution of the latter is actually much less than shown, so that halving the
contribution of the X-chromosome ("dosage compensation") in female generations
would keep the cytosolic protein concentration essentially independent of the sex of the host cell.
[This figure was not included in the original 1994 paper.] |
|
What is it about the intracellular environment that has to
be stabilized? Why is fine-tuning important? A possible answer was given previously when
addressing another paradox (Forsdyke, 1985, 1991,
1992). Cytotoxic T cells recognize
surface complexes of MHC class I proteins in association with peptides derived from
proteins synthesized within the same cell. Since mechanisms of self/not-self
discrimination appear to be extracellular, cells are held to load MHC proteins
intracellularly with peptides from both self
and not-self proteins for display at the cell surface.
Some MHC-self-peptide complexes are
indeed found (Rotzschke & Falk, 1991). However, the virtue of an obligatory display of
self peptides is not readily apparent; it requires the prior deletion or inactivation of
all T cells specific for MHC-self-peptide complexes, thus generating extensive "holes" in the T-cell repertoire
(Du Pasquier & Blomberg,
1982; Vidovik & Matzinger, 1988; Schild et al., 1990; Ohno, 1991). A mechanism
permitting some intracellular discrimination between self proteins and not-self proteins
would allow the preferential loading of MHC class I proteins with peptides derived from
not-self proteins, thus avoiding the logistic problem of competition with a myriad of
peptides derived from self proteins.
For a cell to distinguish intracellularly
between a self protein and a not-self protein (encoded by an intracellular pathogen),
would appear to be a formidable problem. Both classes of protein might, after all, be
synthesized on host ribosomes and might be released into similar cytosolic compartments.
However, there are two key differences which might be exploited by an appropriate
surveillance mechanism.
The first of these is that, relative to not-self protein encoding
genes, each self protein-encoding gene has had more evolutionary time to fine-tune its
product concentration to the intracellular environment created by the other self genes
with which it has been travelling through the generations.
The second is that, whereas self protein-encoding genes are
essentially at peace with the concentrations at which their products have arrived, the raison
d'etre of most foreign pathogens is, at some time within the lifespan of their host,
to increase in number. This may imply synthesizing specific proteins at rates resulting in
unacceptably high cytosolic concentrations.
If a pathogen with a short generation time and high mutation rate could gain a
foothold, it would readily accommodate to the first difference. However, the second
difference, combined with the heat-shock response (discussed elsewhere; Forsdyke, 1994;
1995), could be decisive.
5.
Differential Aggregation of Foreign Proteins
How would an "unacceptably
high" cytosolic concentration be detected? When macromolecules in solution
reach a critical concentration it becomes energetically more favourable for them to
aggregate, like-with-like, than to remain in simple solution. The aggregation involves a
liberation of bound water and an increase in entropy. Being primarily entropy driven, the
aggregation is promoted by an increase in temperature ( Lauffer, 1975, 1989; Leikin &
Parsegian, 1992).
The crowded cytosol constitutes an environment which
readily drives proteins out of solution when they exceed individual concentration
thresholds, This is well recognized from the difficulties encountered when trying to
over-express proteins within foreign cytosols using expression vectors. In such systems
the formation of insoluble aggregates is greatly enhanced by increasing temperature over a
physiological range. This has been shown very clearly in a recent study of foreign protein
expression in mouse cells ( Nguyen et al., 1989).
The proposal, then, is that not-self proteins more
readily "trip" the intracellular surveillance
system because their concentrations are not so fine-tuned to the phase-separating
proclivities of the crowded host cytosol as are the concentrations of self proteins (Fig.
2). In an organism without cytotoxic T cells, or their equivalent, this would trigger cell
death ( Forsdyke, 1995). In an organism with T cells the aggregates would be directed to a
site for degradation to peptides. Specific peptides would then be displayed in association
with MHC class I proteins at the cell surface (Rotzschke & Falk,
1991). A cell would
thus become labelled for destruction by cytotoxic T cells (Forsdyke, 1995). An advantage
of this scheme is that it leaves open for an organism the option of declaring one of its
own self proteins as "foreign" should its synthesis
or turnover become disordered.
|

FIG. 2. Theoretical curves showing that most
gene products only limit phenotype at low concentrations, and that aggregation occurs at
concentrations exceeding that corresponding to two autosomal allelic genes or one
mammalian X-linked gene.
Although gene dosages are normally discrete, gene product
(protein) concentrations (circles) and phenotypes (triangles) are shown as
continuous functions. The situation prevailing in a normal cytosol is indicated by the
vertical arrow. In the absence of aggregation, the concentration of gene product is taken
to be a rectilinear function of the dosage (copy number) of a constitutive gene (filled
circles). Normally, a particular gene product only limits the phenotypic character to
which it contributes at low concentrations. As the normal cytosolic concentration is
approached other gene products become limiting (plateau in values of phenotypic character
assayed; filled triangles).
Thus, decreasing gene product concentration by half may not
change the phenotype. At above normal gene dosages, gene product aggregation occurs and
there is a fall in the quantity of gene product present in non-aggregated form (loss of
rectilinearity; open circles). These changes may have some effect on phenotype (fall in
phenotypic parameter assayed; open triangles), but the major effect will be the
"tripping" of the intracellular not-self surveillance system by aggregates. |
|
6. Role
of X Chromosome Inactivation
Without dosage compensation there would be a
fluctuation in the total intracellular protein concentration between male and female
generations. This would imply a fluctuation in the pressure to drive individual proteins
out of simple solution when their concentrations exceeded specific concentration
thresholds. An increase in the activity of the X chromosome in male generations
(Muller,
1948; Kuroda et al., 1991), or inactivation of one X chromosome in female
generations (Lyon, 1992), would stabilize the pressure and favour fine-tuning of
gene-product concentrations over evolutionary time.
A corollary of this is that, in
addition to being under evolutionary constraint to preserve specific function, genes encoding proteins are also under
evolutionary constraint both to maintain the collective pressure to drive individual
proteins from solution and to maintain individual protein solubilities in the face of that
collective pressure (Forsdyke, 1994b).
A disturbance of phenotype resulting from
hemizygosity enforced by deterioration of the Y chromosome could involve the specific functions of sex chromosome-encoded proteins
and/or their collective functions as
proteins per se. While the concentration of a protein in a heterozygote might
decrease to a level insufficient to affect specific function (i.e. the concentration of
the protein would still correspond to a point on the plateau of the dose-response curve;
Forsdyke, 1994), the decrease in concentration of the protein might itself be sufficient
to affect genetic fitness due to an "exceedingly minute",
but real, influence on some concentration-dependent collective protein function, such as
intracellular self/not-self discrimination. This would have driven the evolution of dosage
compensation and would appear to resolve Muller's paradox (Muller,
1948). Collective
functions less likely to be involved would include effects on the polymerization of
tubulin and actin, and the binding of ions (Donnan equilibrium).
7.
Two Active X Chromosomes in Oocytes and Embryos
No dosage compensation is evident in mammalian oocytes or in female
preblastula embryos (Lyon, 1992).
[But see Added
Note March 2009 at end of Aneuploidy paper.] The prolonged phase of X-chromosome decondensation in
meiotic oocytes probably relates to the need to reactivate the inactive X-chromosome
(resetting methylation patterns), and to correct DNA damage (Bernstein & Bernstein,
1991). This is obviously a very special case for which there should be mechanisms to
prevent inadvertent aggregation of X-encoded proteins.
The preblastula embryo is also a
special case. Even if some intracellular aggregation were to occur and peptide
presentation were possible in a preblastula embryo, at this stage of development the
differentiation of cytotoxic T cells would not have occurred. It is possible that
aggregation of an X-encoded regulatory protein in a female preblastula embryo would play a
critical role in switching off one of the X-chromosomes (Forsdyke,
1995).
8.
Compensation in Z/W Chromosome Systems
In some species (e.g. birds, butterflies, snakes),
the female is the heterogametic sex with Z and W chromosomes and the male is homogametic
(two Z chromosomes). Whereas in eutherian mammals the X-chromosome is about 5% of the
genome, the avian Z-chromosome is about 10%, which approaches the value for the fruit fly.
Thus, if compensation is good for mammals it would seem even more appropriate that
compensation exist in birds.
Dosage compensation in avian systems does not seem to follow
the mammalian model; there is no equivalent of the Barr body and both Z chromosomes
replicate together in males (Ohno, 1967). On the basis of very limited studies it has been
argued that species with Z/W chromosome systems are dosage tolerant (Johnson & Turner,
1979; Baverstock et al., 1982). However, these studies measured protein activities, not actual protein concentrations.
It has been
found that, even in the case of compensated genes, product activity levels may vary
between the sexes in different tissues, presumably in response to hormonal influences
(Steel & Midgeon, 1973). It is predicted that dosage compensation will be found in Z/W
chromosome-bearing species, possibly following the fruit fly model with increased
expression of the Z chromosome when in the heterogametic sex. The reason for this is that
intracellular self/not-self discrimination is a quite fundamental process conferring
resistance to infection and is likely to have evolved well before the evolution of sex
chromosomes (Forsdyke, 1985, 1991, 1992). Other predictions of the hypothesis are
presented elsewhere (Forsdyke, 1995).
|
The author thanks Dr Susamo Ohno for review of an early version
of the manuscript and the Medical Research Council of Canada and the Leukaemia Research
Fund of Toronto for support.
REFERENCES
BAVERSTOCK, P. R., ADAMS, M., POLKINGHORNE, R. W. & GELDER,
M. (1982). A sex-linked enzyme in birds: Z-chromosome conservation but no dosage
compensation. Nature, Lond 296, 763-766.
BERNSTEIN, C. & BERNSTEIN, H. (I 991). Aging, Sex and DNA Repair. New York: Academic Press.
BOOK, J. A. & SANTESSON, B. (1961). Nuclear sex in triploid
XXY human cells. Lancet 2, 318.
BULL, J. J. (1983). Evolution
of Sex-Determining Mechanisms. Menlo Park, CA: Benjamin-Cummings.
CHANDRA, H. S. (1985). Is human X-chromosome inactivation a
sex-determining device? Proc. natn. Acad Sci. U.S.A.
82, 6947-6949.
CHARLESWORTH, B. (1978). Model for evolution of Y-chromosomes
and dosage compensation. Proc. natn. Acad. Sci. U.S.A.
75, 5618-5622.
CHARLESWORTH, B. (1991). The evolution of sex chromosomes. Science 251, 1030-1033.
DU PASQUIER, L. & BLOMBERG, B. (1982). The expression of
antibody diversity in natural and laboratory-made polyploid individuals in the clawed toad
Xenopus. Immunogenetics 15, 251-261.
FORSDYKE, D. R. (1985). Heat shock proteins defend against
intracellular pathogens: a non-immunological basis for self/notself discrimination. J. theor. Biol. 115, 471473.
FORSDYKE, D. R. (1991). Early evolution of MHC polymorphism. J. theor. Biol. 150, 451-456.
FORSDYKE, D. R. (1992). Two signal model of self/not-self immune
discrimination: an update. J. theor. Biol.
154, 109-118.
FORSDYKE, D. R. (1994). The heat-shock response and the
molecular basis of genetic dominance. J. theor. Biol. 167,
1-5.
FORSDYKE, D. R. (1995). Entropy-driven protein self-aggregation
as the basis for self/not-self discrimination in the crowded cytosol: what the Greeks did
not know. J. Biol. Systems 3,
273-287.
HALDANE, J. B. S. (1933). The part played by recurrent mutation
in evolution. Am. Nat. 67, 5-19.
HARNDEN, D. G. (1961). Nuclear sex in triploid XXY human cells. Lancet 2, 488.
HODGKIN, J. (I 990). Sex determination compared in Drosophila
and Caenorhabditis. Nature, Lond. 344,
721 728.
JACOBS, P. A. & MIDGEON, B. R. (1989). Studies of
X-chromosome inactivation in trisomies. Cytogenet. Cell
Genet. 50, 75-77.
JOHNSON, M. S. & TURNER, J. R. G. (1979). Absence of dosage
compensation for a sex-linked enzyme in butterflies (Heliconius). Heredity 43, 71-77.
KURODA, M. I., KERNAN, M. J., KREBER, R.,
GANETZKY, B. & BAKER, B. S. (1991). The maleless protein associates with
the X-chromosome to regulate dosage-compensation in Drosophila. Cell 66, 935-947.
LAUFFER, M. A. (1975). Entropy-driven
Processes in Biology. New York: Springer-Verlag.
LAUFFER, M. A. (1989). Motion
in Biological Systems. New York: A. R. Liss.
LEIKIN, S. & PARSEGIAN, V. A. (1992). A
mechanism for temperature-favoured protein assembly. FASEB
J. 6, 418.
LINDSLEY, D. L., SANDLER, L., BAKER, B. S.,
CARPENTER, A.T., DENELL, R. E., HALL, J. C., JACOBS, P. A.,
MIKLOS, G. L., DAVIS, B. K., GETHMAN, R. C., HARDY, R.W., HESSLER,
A., MILLER, S.M., NOZAWA, H., PARRY, D. M. & GOULD-SOMERO, M.
(1972). Segmental aneuploidy and the genetic gross structure of the Drosophila genome.
Genetics 71, 157-184.
LYON, M. F. (1992). Some milestones in the history of
X-chromosome inactivation. A. Rev. Genet.
26, 17-28.
MULLER, H. J. (1948). Evidence on the precision of
genetic adaptation. Harvey Lect. 43,
165 229.
NGUYEN, V. T., MORANGE, M. & BENSAUDE, 0. (1989).
Protein denaturation during heat-shock and related stress. E. coli. beta-galactosidase
and Photinus pyralis luciferase inactivation in mouse cells. J. Biol. Chem. 264, 10487-10492.
OHNO, S. (1967). Sex
Chromosomes and Sex-Linked Genes. New York: Springer-Verlag.
OHNO, S. (1973). Conservation of ancient linkage groups in
evolution and some insights into the genetic regulatory mechanisms of X-inactivation. Cold Spring Harb. Symp. Quant. Biol. 38,
155-164.
OHNO, S. (1991). To be or not to be a responder in T-cell
responses: ubiquitous oligopeptides in all proteins. Immunogenetics
34, 215-221.
ORR, H. A. (1990). "Why polyploidy is rarer in
animals than plants" revisited. Am. Nat.
136, 759 770.
ROTZSCHKE, 0. & FALK, K. (1991). Naturally occurring
peptide antigens derived from the MHC class I-restricted processing pathway. Immun. Today, 12, 447 455.
SCHILD, H., ROTZSCHKE, O., KALBACHER, H. &
RAMMENSEE, H. G. (1990). Limit of T-cell tolerance to self proteins by peptide
presentation. Science 247, 1587
1589.
STEEL, M. W. & MIDGEON, B. R. (1973). Sex
differences in activity of glucose-6-phosphate dehydrogenase of cultured human foetal lung
cells despite X-inactivation. Biochem. Genet.
9, 163-170.
STEINEMANN, M. (1982). Multiple sex chromosomes in Drosophila
miranda: a system to study the degeneration of a chromosome. Chromosoma 86, 59-76.
VIDOVIK, D. & MATZINGER, P. (1988).
Unresponsiveness to a foreign antigen can be caused by self-tolerance. Nature, Land. 336, 222 -225.
 [ADDED NOTE:
2001. Strong evidence
for avian dosage compensation was reported by McQueen, H., McBride, D., Miele,
G., Bird, A. P., & Clinton, M. (2001) Dosage compensation in birds. Current
Biology 11, 253-257.
[ADDED NOTE:
2001. Strong evidence
for avian dosage compensation was reported by McQueen, H., McBride, D., Miele,
G., Bird, A. P., & Clinton, M. (2001) Dosage compensation in birds. Current
Biology 11, 253-257.
ADDED NOTE: 2005. See also
Cole, L. J. & Hollander, W. F. (1950) Hybrids of pigeon by ring dove. American
Naturalist 84,
275-307. In private correspondence with A. G. Cock (May
20th 1968) Hollander writes:
"For one locus in the pigeon and the dove (Streptopelia
risoria) we have a graded series of recessive mutant alleles which I
think meet your strict requirements.... The alleles d and
"blond" of pigeon and dove, respectively, are homologous as
shown by a hybrid test. "White" (= extreme dilution) in the
dove is a lower allele, and the heterozygous blond/white male is paler
than homozygous blond. Both in pigeon and dove the phenotypes of dilute
= blond females and homozygous males are indistinguishable. The example
is in my opinion a clear case of dosage compensation (sensu
strictu), but it has not been emphasized as such in the literature." |
[This is in the AGC
papers, deposited in 2008 in Queen's University Archives
Click Here
]
ADDED NOTE:
May 2008.
| Failure to inactivate an X chromosome
would disrupt the fine-tuning of intracellular protein concentrations
and could predispose females to autoimmune disease (see H. F. Pan et al.
2008 Medical Hypothesis 70,
1231-1232; D. L. Smith-Bouvier et al. 2008 J.
Exp. Med. 205, 1099-1108.). |
ADDED NOTE:
Sept 2008.
Following the full sequencing of two
avian genomes, further evidence for avian dosage compensation is
summarized by Arnold, Itoh and Melamed (2008 Annual
Review of Genomics and Human Genetics 9, 109-127).
Unlike most previous reviews, which dwell on mechanisms (the
"how" question), the authors discuss the selective pressures
that might have brought about the evolution of dosage compensation (the
"why" question). However, unaware of the hypothesis advanced
here, they provide an ad hoc "network" explanation in terms of
critical "hub genes" that regulate a multiplicity of other
genes. They rightly point to the fact that dosage compensation is not
normally brought about by variations in copy number of individual genes,
or even of small chromosomes, but
then continue:
"In contrast, monosomy or
trisomy for a whole large
chromosome usually presents a major problem; in humans, for
example, either is usually lethal. This is probably because
differences over a large genomic segment are more likely to
involve differences in dose of critical hub genes. The more hub
genes involved, the larger the problem. ... This view suggests
that the reduction in dose of a minority of X genes drove the
selection of complex molecular mechanisms to adjust X dosage in
one sex or the other." |
|
ADDED
NOTE: Nov 2008
| H-F. Pan and his
colleagues further marshal the evidence for failure to completely
inactivate one of the X chromosomes in SLE in a new article (Medical
Hypothesis 72, 99-109). The fact that failure to
inactivate an X chromosome in females would increase the aggregation
pressure is now acknowledged: "In these
females a higher than normal aggregation pressure would be expected." |
3. Aneuploid Lethality
Fine Tuning of
Intracellular Protein Concentrations, a Collective Protein Function Involved in Aneuploid Lethality, Sex-Determination and Speciation?
Journal of Theoretical Biology (1995) 172,
335-345.
(With copyright permission from Academic Press)
(Received on 26 January 1994, Accepted in revised form on 24 August 1994)
Abstract
1. Introduction
2. Aneuploid Lethality
2.1.
CUMULATIVE LOSS/GAIN OF MANY SEGMENTS REQUIRED FOR LETHALITY
2.2.
MANY CHROMOSOMAL ELEMENTS DETERMINE SEX AND DOSAGE COMPENSATION
2.3. X-CHROMOSOME DOSAGE
COMPENSATION
3. Aggregation Hypothesis
3.1.
SOME MUTATIONS AFFECT SPECIFIC PROTEIN CONCENTRATION, NOT FUNCTION
3.2.
FINE-TUNING OF INTRACELLULAR PROTEIN CONCENTRATIONS TO A MAXIMUM CONSISTENT WITH AVOIDING SELF-AGGREGATION
3.3. DOSAGE
COMPENSATION NECESSARY FOR FINE-TUNING
3.4. CELL
VOLUME AND FINE-TUNING IN DIFFERENT TISSUES
4. Implications of Hypothesis
4. 1.
TEMPERATURE SELECTIVELY DESTROYS MALE INTER-SEX CELLS
4.2.
DEPENDENCE ON X/A RATIO MEANS DEPENDENCE ON CONCENTRATION OF X-ENCODED PRODUCTS
4.3.
LESS AGGREGATION PRESSURE IN MALE ZYGOTES SWITCHES ON X HYPERTRANSCRIPTION
4.4.
SPECIATION AS A COLLECTIVE GENE FUNCTION. HALDANE'S RULE
4.5.
RECOMBINATION HELPS GENES PREDICT THEIR FUTURE ENVIRONMENT
4.6. FINE-TUNING IN ASEXUAL
SPECIES
End Note (Oct. 2008)
End Note (March 2009)
End_Note_(Oct_2010)
Abstract.
The assertion that sex-chromosome dosage compensation arose because aneuploidy for an
entire chromosome is lethal, begs the question of why aneuploidy is lethal. It has been
proposed that aneuploid lethality results from impairment of a collective protein function
(Forsdyke, 1994, J. theor. Biol. 167,
7-12).
Cytosolic
proteins, by virtue of their concentrations, exert a pressure tending to drive members of
individual protein species into self-aggregates. Other evolutionary time, each gene has
fine-tuned the concentration of its product to a maximum consistent with avoiding
self-aggregation in the crowded cytosol.
Because of this aggregation pressure and the
imprecision of their own fine-tuning, the proteins of members of other species, the
corresponding genes of which may have been transported to a cell as viruses (or gametes),
are specifically aggregated. The death of the cell and its enclosed virus results.
Aneuploidy impairs this process, with lethal consequences for the organism.
The hypothesis
leads to explanations for a variety of phenomena.
On the assumption that the concentration
of autosomal products determines cell volume, the observed dependence of sex-determination
on the ratio of X-chromosomes to autosomes is shown merely to be a dependence on the
concentration of the products of one X-chromosome.
The inviability of the heterogametic
sex among the offspring of an interspecies cross (Haldane's rule), follows from the
species-specific fine-tuning of the concentrations of X-chromosome encoded
products, relative to the concentration of autosomally encoded products.
|
|
1. Introduction
In species with sex chromosomes, one sex is usually
haploid with respect to one of the sex chromosomes. Thus, in humans and fruit flies, males
have only one copy of the X-chromosome ( Jablonka & Lamb, 1990;
Charlesworth, 1991).
Since the concentration of a final gene product (usually a protein) is often directly
related to the number of gene copies, the concentration of X-chromosome gene products in
males should be half that in females. The process known as dosage compensation ensures
that this does not occur. In humans dosage compensation is achieved by inactivating one of
the X-chromosomes in females (Lyon, 1992). In the fruit fly Drosophila melanogaster
the transcriptional activity of the single male X-chromosome is increased (Muller, 1948;
Stern, 1960).
Muller's choice of the
value-laden
term "compensation" to describe the adjustment of
dosage implies that the process is advantageous. What adaptive advantage could dosage
compensation confer? The standard answer to this question begins by noting that, whichever
chromosome is affected, aneuploidy for more than 3% of a genome is usually
lethal. Since
the human X-chromosome is approximately 5% of the genome and the fruit fly X-chromosome is
approximately 20% of the genome, then without dosage compensation there might be an
intolerable degree of aneuploidy.
This answer was given by Muller & Kaplan
(1966), and
by Ohno (1967), and has been repeated in most subsequent reviews
(e.g. Baker & Belote,
1983; Jaffe & Laird, 1986). Certainly, mutations in genes affecting dosage
compensation are usually lethal (Lucchesi & Manning, 1987; Hodgkin,
1990). This
implies that aneuploidy for the X-chromosome would indeed be lethal. However, why aneuploid states are lethal is not explained.
If one accepts the deduction that X-chromosome
aneuploid lethality is just one example of the general problem of aneuploid lethality,
then two inductions can be made:
(i) Since X-chromosome aneuploidy in male fruit flies and humans can
be compensated by adjusting the concentrations of X-encoded gene products, then aneuploid
states, in general, may be lethal because of disturbances at the gene product level.
(ii) Since most of the X-chromosome is the subject of compensation,
then aneuploid states, in general, may be lethal because of a collective disturbance of
many
gene products. Thus, aneuploid lethality is
unlikely to occur because there is a small subset of genes which are aneuploid sensitive.
In this paper I first review evidence supporting these
inductions, derived from studies in three areas;
-
(i) segmental aneuploidy,
-
(ii)
determination of sex by the ratio of X-chromosomes to autosomes (X/A ratio) and
-
(iii) the
evolution of dosage compensation in the apparent absence of any selective advantage
(Forsdyke,
1994b).
I then review a recently proposed mechanism by which gene products
could act collectively to produce lethal effects (Forsdyke,
1995). Finally, some
implications for the chromosomal determination of sex, and interspecies incompatibility at
the intracellular level, are discussed.
2.
Aneuploid Lethality
2.1. CUMULATIVE LOSS/GAIN OF MANY
SEGMENTS REQUIRED FOR LETHALITY
Lindsley and co-workers
( 1972) studied segmental
aneuploidy in fruit flies. Small chromosome segments were either deleted to make a region
haploid, or duplicated to make the region triploid. Aneuploidy for only one locus was
found to be lethal. This was the triplolethal locus, which appears to encode an RNA
helicase (Dorer et al., 1990). In this case, neither the haploid state nor the
triploid state was tolerated. This would explain why the loss or gain of a DNA segment
containing triplolethal would not be accepted, but it would not explain aneuploidy
as a general phenomenon. In general, segments could be removed or duplicated with no
serious impairment of function. A collective
loss/gain of many segments was
necessary for lethality. Lindsley and co-workers concluded that:
"The deleterious effects of aneuploidy are, in the main,
caused by the additive
effects of genes that slightly reduce
viability, and not by the individual effects of a few aneuploid-lethal genes among a large
array of dosage-insensitive loci" ( my
italics).
2.2. MANY CHROMOSOMAL ELEMENTS DETERMINE SEX AND DOSAGE
COMPENSATION
Another line of evidence in support of the above
inductions derives from studies of the determination of sex and dosage compensation in
certain polyploid variant organisms. These studies show that sex chromosome dosage
compensation depends, not on the absolute number of X-chromosomes, but on the X/A ratio.
In fruit flies, both sex and dosage compensation are determined by the X/A ratio. In
humans, only dosage compensation is determined by the X/A ratio ( Book &
Santesson,
1961; Harnden, 1961; Mittwoch, 1973; Webb et al., 1992).
In 1983 Baker & Belote concluded that the X/A
ratio "remains the most enigmatic aspect of the hierarchy
controlling sex and dosage compensation". In spite of dramatic advances in our
understanding of the molecular basis of the implementation
of the information provided by the X/A ratio, a decade later the enigma remains. It
has been argued that a limited number of X-encoded proteins, (such as sis-a,
runt and the
helix-loop-helix protein sis-b), may be sufficient to act as numerators,
and that a limited number of autosomally encoded proteins (such as the helix-loop-helix
protein deadpan) may be sufficient to act as denominators
( reviewed by McKeown & Madigan,
1992). These arguments do not take into account the
historical evidence for a great multiplicity of numerator/denominator
elements.
Dependence of sex determination on the X/A ratio has
been described for the fruit fly ( Bridges,
1925).
-
A ratio equal to or less than 0.5
generates a male (chromosome complement designated X,2A if the Y-chromosome is ignored).
-
A
ratio equal to or more than 1.0 generates a female (2X,2A).
-
A triploid individual (3X,3A)
has a ratio of 1.0 and is female.
-
A triploid individual with only one X-chromosome
(1X,3A)
has a ratio of 0.33 and is male.
-
A triploid individual with two X-chromosomes
(2X,3A),
however, has a ratio of 0.67. Such "intersexes"
have varying proportions of male and female cells, each cell type being found in course
patches indicating an early random commitment of individual cells to become clones of a
particular sex (Sanchez & Nöthiger,
1983).
2.2.1. Numerator elements
The addition of small segments of the X-chromosome to
2X,3A zygotes, prior to the time the male/female decision is made, should increase the
proportion of female cells. This was indeed found ( Dobzhansky & Schultz, 1934; Baker
& Belote, 1983). Two remarkable features of these experiments were that:
(i) Most small segments of the X-chromosome were
equal
with respect to their abilities to increase the
proportion of female cells.
(ii) Increasing the length of the segment increased the proportion of female cells.
Thus the numerator element in the
X/A ratio appeared as a collective function
of X-chromosome segments and, by implication, of their genes and gene products.
2.2.2. Denominator elements
Baker & Belote
( 1983) were able to conclude only that:
"The autosomal [ denominator] component(s) whose dosage is
important for the primary determination of sex and/or dosage compensation has eluded
detection".
Little progress has been made in subsequent fruit fly studies
( Younger-Shepherd et
al., 1992). However, studies in human trisomies provide some evidence that the
denominator element in the X/A ratio determining dosage compensation might also be
a
collective function (Jacobs & Midgeon,
1989).
In normal human males (designated
X,2A if we ignore
the Y-chromosome), there is one active X-chromosome which [ for
convenience] can be
regarded, by analogy with the fruit fly, to be permanently "hypertranscribed"
[relative to those of the female Xs if the latter were fruit fly
Xs], so that its products
"balance" those of the autosomes. Thus, the ratio
of X-encoded products to autosome-encoded products can be designated as 1.0.
In normal human females
(2X,2A) one X is inactive and replicates late in S-phase. The
other X is "hypertranscribed" as in the male
(giving a product ratio of 1.0). In tetraploid females (4X,4A) there are
two late-replicating X-chromosomes and two "hypertranscribed"
X-chromosomes whose products would thus balance those of the four sets of autosomes
(product ratio again 1.0).
In triploids
(3X,3A) there are two cell populations
reflecting two options, neither of them perfect. If there are two "hyperactive" X-chromosomes (and one inactive) then the level
of X-chromosome products is set to the level required to balance the products of four, not
three autosomes (predicted product ratio 1.33). If there is one "hyperactive" X-chromosome (and two inactive) then the level of
X-chromosome products is set to the level required to balance the products of two, not
three autosomes (predicted product ratio 0.67).
Jacobs and Midgeon asked whether there is one
powerful denominator autosome, or whether the denominator function is a collective
function of more than one autosome. They found, in individual human females, each trisomic
for a particular autosome, no cells in which the presence of the extra autosome would
allow both X-chromosomes to remain active. The study was unsatisfactory for various
reasons, particularly the fact that spontaneous trisomies for four of the 22 possible
autosomes were not found. Nevertheless, the data were consistent with the hypothesis that
the denominator function is collective. The authors wondered whether compensation
involves:
". . the action of a specific autosomal gene(s), or does it
depend on a more indirect attribute,
such as an increase in
nuclear volume ... ?" (my
italics).
In the same vein, Dobzhansky and Schulz wrote in 1934:
"One may, perhaps, account for the sex-determining function
of the autosomes even without assuming any autosomal sex genes. Sex may be determined by
the ratio between the number of X-chromosomes present in the cell and the size of the
cell. Cell size in most organisms (including Drosophila), is positively correlated
with the volume of chromosomal material contained in the nucleus."
It should be noted that hyperploidy is usually associated with an increase in cell volume
(Dobzhansky, 1929; Tal, 1980;
Lucchesi & Manning, 1987), and hypoploidy with a decrease
in cell volume (Bridges, 1939). This would appear to reflect the general dependence of
protein quantity on gene dosage. A change in volume would accommodate a change in the
quantity of an intracellular protein while maintaining its concentration constant
(Colclasure & Parker, 1991). This is an important point to which we will be returning.
2.3.
X-CHROMOSOME DOSAGE COMPENSATION
Many genes encode proteins with dosage sensitive
functions, such as enzymes. Typically, with increasing enzyme concentration there is a
progressive increase in the quantity of enzyme product formed. The quantity of this
product may be reflected in the phenotype. However, above a certain enzyme concentration
some other factor becomes rate-limiting (e.g. substrate availability); curves plotting the
rate of product formation as a function of enzyme concentration then tend to plateau.
2.3. 1. Dosage imbalance does not impair
specific protein functions
Muller (1948) was struck by the fact that
the
concentrations of most enzymes found within diploid cells correspond to a point well along
the plateau of the dose-response curve. In most cases, halving the concentration of the
enzyme would still generate a product level corresponding to the plateau of the
dose-response curve ( Forsdyke,
1994a). Thus, there would be no phenotypic difference
between a female fruit fly cell containing two active X-chromosomes and a male fruit fly
cell containing one uncompensated
X-chromosome. Muller's paradox is that dosage compensation has evolved when there would
appear to be only, in his words, "exceedingly minute
differences" between compensated and uncompensated phenotypes.
2.3.2 Dosage imbalance impairs collective protein
function?
It has been proposed that the paradox would be
resolved if the pressure to evolve dosage compensation involved selection acting, not on
the unique individual phenotypes characteristic of each individual gene product, but on a
phenotype generated collectively by all X-encoded gene products ( Forsdyke,
1994b).
The
Donnan equilibrium is an example of a collective protein function. Intracellular proteins
at physiological pH are usually charged molecules which can influence the equilibrium
distribution of ions across protein-impermeable membranes (Hitchcock,
1924). Over the
range of protein concentrations found in cells, the strength of the Donnan effect is
directly proportional to the quantity of intracellular protein. There is not an
intracellular protein concentration beyond which the effect no longer increases. If the
X-chromosome (20% of the genome) were not hyperactivated in the male fruit fly, then male
cytoplasm would have approximately 90% of the protein concentration of the female
cytoplasm. A difference of this magnitude is not "exceedingly
minute". It might adversely affect the Donnan equilibrium and hence
negatively influence phenotype.
However, the Donnan effect would appear to be
volume-sensitive. Adjusting cell volume should correct a concentration difference between
proteins in male and female cells. That this may not be feasible becomes apparent on
considering a novel collective function of intracellular proteins -- this is discussed
below.
3.
Aggregation Hypothesis
3.1. SOME MUTATIONS AFFECT SPECIFIC PROTEIN CONCENTRATION, NOT
FUNCTION
Some gene mutations affect the functions of unique
gene products. Other mutations affect the concentrations of those products, often without
influencing function per protein molecule. For example, the concentration of a protein
might be changed if the concentration of its mRNA were changed, either by a promoter
mutation affecting the transcription rate, or by a mutation in the 3' non-coding region
affecting mRNA stability. Similarly, a mutation in a protein encoding region might affect
protein stability, but have no effect on function per protein molecule.
As discussed above, within cells the intracellular
concentration of most proteins corresponds to a point well along the plateau of the plot
of activity (phenotype) against protein concentration. When mutations occur resulting in a
specific decrease in concentration of
a protein, the protein concentration will move to the left, corresponding to points on the
plateau of the dose-response curve.
Eventually a point will be reached where specific
function will begin to be impaired. There will then be an effect on phenotype and a
decrease in fitness. A detailed discussion of this, in the context of the evolution of
genetic dominance, is presented elsewhere (Forsdyke,
1994a). The basic point being made
here is that there is a lower limit below which a mutation-driven decrease in
concentration of a gene product cannot occur without affecting specific function.
By the same token, when mutations which specifically
increase the concentration of a
particular protein occur, the protein concentration will move to the right corresponding
to points on the plateau of the dose-response curve. In this case, there is no obvious
change in specific phenotype. For a given intracellular protein, does the actual
concentration found among members of a biological species fluctuate between the lower
limit, defined above, and an upper limit set simply by the solution limit? Alternatively,
are there further selective forces which determine a precise concentration setting for
each protein?
3.2. FINE-TUNING OF INTRACELLULAR PROTEIN CONCENTRATIONS TO A
MAXIMUM CONSISTENT WITH AVOIDING SELF-AGGREGATION
Generally, it is easier for a protein to remain
soluble in a salt solution at physiological pH than to remain soluble in the "crowded cytosol"
(Fulton, 1982). The solubility of a
particular protein species is determined by the concentrations of the other proteins that
accompany it (Lauffer, 1975). It has been proposed that, over evolutionary time, each
individual gene fine-tunes the concentration of its product to the concentrations of the
products of the other genes with which it has been travelling through the generations
(Forsdyke, 1995).
Irrespective of its specific function, the "aim"
of each gene is to maximize the concentration of its own products. Collectively, the
products of all genes exert a pressure tending to drive individual protein species into
aggregates if they exceed their solubility limit. Each individual protein species both
contributes to the pressure and is acted upon by the pressure (Fig. 1).
|
| FIG. 1. What point on the dose-response curve
corresponds to the normal concentration of a gene product within a cell?
This hypothetical
in vivo dose-response curve shows some quantitative measure of phenotype (e.g. rate
of formation of enzyme product) as a function of the concentration of a gene product which
contributes to that phenotype (such as the concentration of an enzyme). The phenotypic
parameter increases with gene dosage until point A when some other factor (e.g.
substrate availability) becomes rate-limiting. The curve then plateaus. B
corresponds to the minimum concentration of gene product required for a "margin
of safety", so that heterozygote function is not impaired (Forsdyke, 1994a).
E corresponds to the concentration at which the gene product would still be soluble
if no other proteins were present. Above this concentration, the protein would
self-aggregate and the phenotypic parameter would decrease. D corresponds to the
concentration at which aggregation would occur in the presence of cytosolic proteins
(Forsdyke, 1995). The horizontal arrows symbolize the aggregation pressure exerted
collectively by cytosolic proteins, which tends to push the descending limb of the
dose-response curve to the left.
Thus, it would seem that the concentration of a protein
in cells of different members of a species could fluctuate between points B and D
with no effect on phenotype. It is proposed, however, that over evolutionary time genes
have "fine-tuned" the concentrations of their products to a
maximum consistent with avoiding self-aggregation. This point might correspond to C
(marked by a vertical arrow), which is slightly to the left of D, thus providing a
small "margin of safety" against inadvertent self-aggregation. |
|
The selective force leading to the evolution of this
pressure involves the invasion of the intracellular space by a member of another species
of organism (e.g. a virus), which has not had the opportunity to fine-tune so precisely
the concentrations of its gene products. As discussed elsewhere (Forsdyke, 1985, 1991,
1995), by mechanisms involving heat-shock proteins, proteins encoded by virus genes would
tend to be specifically aggregated. Irrespective of any effect on specific function, this
aggregation would trip an output system which, depending on the organism, would involve
cell death inflicted either directly, by an intracellular mechanism (e.g. apoptosis), or
indirectly, by cytotoxic T cells. A key feature of the aggregation is that it is primarily
entropy-driven and thus is favoured by a small increase in temperature (Lauffer,
1975).
The death of the cell and the pathogen it contains would usually be beneficial to the
organism. In some cases aggregation alone might suffice to limit the spread of the
pathogen.
3.3. DOSAGE COMPENSATION NECESSARY FOR FINE-TUNING
The need for fine-tuning over evolutionary time
provides a clear rationale for the evolution of dosage compensation (Forsdyke,
1994b).
Without dosage compensation, the cytosolic protein concentration would fluctuate between male and female generations.
Fine-tuning would be possible only for genes encoded by Y-chromosomes, which are eternally
locked in male generations. Failure to fine-tune would impair intracellular self/not-self
discrimination and thus increase the vulnerability of an organism to intracellular
pathogens.
|
| FIG. 2.
Evolutionary fine-tuning of intracellular protein concentrations as an explanation for
X-chromosome dosage compensation.
Rectangles refer to maternal (M) and paternal (P)
haploid chromosome complements which combine to form either (a) a female zygote, (b) an
uncompensated male zygote, or (c) a compensated male zygote. Gene products (proteins) are A,
B and C (for the X-chromosome), D, E and F (for
autosome A1), and G and H (for autosome A2). The specific function of these proteins is not relevant to the
primary establishment of the X/A signal governing the initiation of X hypertranscription
in male fruit fly (dosage compensation).
The numbers below each protein indicate its
quantity, which has been fine-tuned over evolutionary time to a maximum consistent with
remaining in solution in the crowded cytosol. Numbers in brackets are the quantities of
products contributed by the maternal X-chromosome, which balance the products of the
paternal autosomes.
The proteins, by virtue of their concentrations, exert a collective
pressure (which can be expressed as the sum of individual protein concentrations), tending
to drive individual protein species into self-aggregates. For female zygotes and
compensated male zygotes the net aggregation pressure might be 40 units
per cell. For an uncompensated male zygote the net aggregation pressure would be only 34
units/cell.
Cell volume cannot be decreased by a factor of 34/40 in male
generations to maintain the aggregation pressure because the quantities of autosomal
products (D-H) are already fine-tuned to a maximum consistent with remaining
soluble in female generations. Since a high aggregation pressure is needed for the
detection of proteins encoded by intracellular pathogens, the viability of uncompensated
phenotypes will be decreased. |
|
Figure 2 shows a model of how this might work. It is
postulated that, without dosage compensation, the discrepancy in cytosolic protein
concentrations between male and female generations would not readily be corrected by an
intergenerational adjustment in cell volume. Reducing cell volume in male generations
[by
a factor of 34/40 in Fig. 2(b)], would slightly concentrate the products
of autosomal genes. Since these had already been fine-tuned in female generations to
maximize the solubility of each individual protein species, a further concentration might
trigger their aggregation. Intergenerational volume adjustment would work only by
compromising the degree of fine-tuning in female generations.
It can be argued that the lower concentration of
X-encoded products would slightly decrease the net pressure tending to drive the
autosomally encoded proteins into aggregates, and this might make it possible to
accommodate autosomal proteins in a smaller volume without triggering their aggregation.
However, it is postulated here that this would not be a feasible alternative. Cell volume
is autosomally determined. Experimental evidence to the contrary would falsify this aspect
of the hypothesis.
3.4. CELL VOLUME AND FINE-TUNING IN DIFFERENT TISSUES
It is apparent that the general setting of cell
volume as a function of the number of chromosomes (see Section
2.2.2) is subject to
tissue-specific modification. The major cell of vertebrate lymphoid tissues, the small
lymphocyte, is an extreme example. The ratio of cytoplasm to nucleus is very low. This can
be regarded as an adaptation for circulating in the lymphatic and vascular systems from
which lymphocytes migrate to regions of inflammation. It can be assumed that the concentration of cytoplasmic proteins is no less than
in a cell with more cytoplasm.
It should also be noted that the differential gene
expression associated with cell specialization requires fine-tuning to a particular subset
of genes in each tissue, as well as to the more generally expressed "house-keeping" genes. This might require different final
concentrations of the same gene product in different tissues. In some cases this could
have been achieved by gene duplication so that the same protein could be expressed under
the influence of different promoters in different tissues.
As discussed elsewhere
(Forsdyke, 1995), it is
envisaged that, in the case of proteins whose concentrations fluctuate as part of
physiological processes, concentration fine-tuning would have been with respect to the
maximum concentration attained. Similarly, in the case of proteins that form
stoichiometric complexes with other proteins, the whole complex would have been treated as
a unit during evolutionary fine-tuning. In the case of reversibly modified proteins
(phosphorylation, glycosylation), concentration fine-tuning would be with respect to the
least soluble form of the protein.
4.
Implications of Hypothesis
4. 1. TEMPERATURE SELECTIVELY DESTROYS MALE INTER-SEX CELLS
The aggregation hypothesis makes numerous explicit
experimental predictions and reinterpretations of previous data ( Forsdyke,
1995). One of
these is that male cells in fruit fly intersexes should be more vulnerable to high
temperatures than female cells in fruit fly intersexes. Cells of a triploid intersex fruit
fly (2X,3A) can have their X-chromosome transcribed either in the male mode (giving a
protein level which would balance that corresponding to four autosomes), or in the female
mode (giving a protein level which would balance that corresponding to two autosomes). The
relative X/A product ratios in each actual case would be 1.33 (male cell)
and 0.67 (female cell).
The excess of X-encoded products in male cells
would tend to aggregate, leading to the selective death of male cells. Since aggregation
is promoted by a small increase in temperature (Lauffer, 1975; Forsdyke,
1994a),
incubating triploid intersexes at high temperatures would enhance the death of male cells,
while having little effect on female cells in which the pressure to aggregate would be
below normal (normal corresponding to a relative product ratio of 1.0).
Evidence in support of this has been presented by Dobzhansky (1930) and Lauge
(1980).
4.2. DEPENDENCE ON X/A RATIO MEANS DEPENDENCE ON CONCENTRATION OF
X-ENCODED PRODUCTS
It was proposed above (Section 3.3) that the volume of a cell is directly related to the
quantity of autosomal products it
must contain. Assuming, for simplicity, a rectilinear relationship, cell volume
(V) is
equal to kAp, where
Ap is the quantity of autosomal products and
k is a constant. It
follows that the concentration of
X-encoded products (Xp /V where
Xp is the quantity of X-chromosome-encoded products), is proportional
to the ratio of the quantity of X-chromosome encoded products to the quantity of
autosomally encoded products. Thus:
Xp/V = Xp/kAp = (1/k)Xp/Ap.
In that the quantities of the products are proportional to the relative
gene dosages, it follows that the concentration of X-encoded products is proportional to
the X/A ratio (defined as the ratio of the number of X-chromosomes to the number of
autosomes). From this it can be concluded that the statement that the determination of sex
and/or dosage compensation is dependent on the X/A ratio (Bridges,
1925), may merely be
acknowledging a dependence on the concentration
of X-encoded products (Xp/V).
4.3. LESS AGGREGATION PRESSURE IN MALE ZYGOTES SWITCHES ON X
HYPERTRANSCRIPTION
Prior to the onset of dosage compensation, male
fruit-fly zygotes would experience a decline in aggregation pressure as X-encoded maternal
gene products decreased to be replaced by a zygote's own single X-encoded products
(Mahowold & Kambysellis,
1980). In female fruit fly zygotes the aggregation pressure
would be sustained because decreasing maternal X-encoded products would be replaced by the
products of both X-chromosomes of the zygote. The aggregation pressure in female cells
(Pf) can be
regarded as equal to k'[2Xc+Ac], where
Ac is the concentration of autosomal products,
Xc is the
contribution to the concentration of X-chromosome encoded products of one X-chromosome and
k' is a constant.
Similarly, the aggregation pressure in uncompensated
male cells (Pm) would be equal to
k'[Xc + Ac]. Then the difference in aggregation
pressure between males and females (Pf
- Pm) would be equal to k'Xc.
The latter includes the concentration of the extra set of X-chromosome encoded products
which, as argued in Section 3.3, would depend on the X/A ratio (since cell volume is held
to be autosomally determined). Thus, the switch to X hypertranscription would depend primarily on the collective
functioning of all X-chromosome encoded genes.
A secondary
effect of the greater pressure in female cells could be that the homo- or hetero-dimerization of helix-loop-helix sex-determining proteins, such as
daughterless
(maternally derived) and sis-b (zygotically-derived), would be promoted. The regulatory
activity of helix-loop-helix proteins is usually greatly affected by such aggregation
(Thayer & Weintraub, 1993). The helix-loop-helix protein aggregates might activate the
zygotic sex-lethal gene early promoter (carried by both X-chromosomes) thus
switching to the female pattern of development (Keyes et al.,
1992). In male
zygotes, the aggregation pressure would be less and helix-loop-helix protein aggregates
would tend to dissociate so that the single sex-lethal gene would be inactive. This
would remove the negative influence of the sex-lethal product on the autosomally
encoded genes which initiate hypertranscription of the male X-chromosome (Gorman et
al., 1993).
4.4. SPECIATION AS A COLLECTIVE GENE FUNCTION. HALDANE'S RULE
A biological species can be viewed as having set up
intracellular barriers against members of other species, be they intracellular pathogens,
or gametes of a species related enough to show no prezygotic isolation (Templeton, 1989;
Coyne, 1992). The fine-tuning of cytosolic protein concentrations, continuing through the
generations and communicated to all interbreeding members of the species, would have
created an intracellular environment hostile to the gene products both of foreign
pathogens, and of the gametes of other species (Forsdyke,
1995).
A failure of members of a species to interbreed,
owing to some form of allopatric or sympatric isolation, would provide an opportunity for
mutational differences to arise between alleles that might affect the abilities of their
products to contribute to the cytosolic aggregation pressure. This might occur before any
allele-specific differences in phenotype could arise. Fusion of gametes from such
reproductively isolated lines would expose a cytoplasmic incompatibility owing to
differences in the fine-tuning of cytosolic protein concentrations. Thus, the list of
potential cytoplasmic incompatibilities between organisms should include "warfare", not only between organelles, as is widely recognized
(Hurst, 1993), but also between cytosolic proteins
(Forsdyke, 1995). The latter might have
played a role in the origin of species.
The particular vulnerability of members of the
heterogametic sex among the offspring of interspecies hybridizations (Haldane's rule), may
serve to illustrate the latter point. The cytosolic concentrations of proteins encoded by
one X-chromosome are uniquely fine-tuned to the concentrations of proteins encoded by its
haploid autosomal complement. In female fruit fly, each X is accompanied by at least one
compatible autosome complement. In male fruit fly, the solitary X is hypertranscribed and
only one X is accompanied by a compatible autosomal complement.
This could result in
changes in net aggregation pressure with adverse consequences for the organism. Figure 3
shows a simple model of how this might work. Any
X-linked gene which mutated so that the concentration of its product changed would suffice
to begin the isolation process. There would be no distinct "speciation
genes".
|
FIG. 3. Evolutionary fine-tuning of gene product
concentrations as an explanation for decreased viability of the heterogametic sex among
the offspring of interspecies crosses (Haldane's rule).
Following the convention of Fig.
2, at the top are the genomes of two wild-type members of a species. During
gametogenesis,
one produces a concentration deficiency mutant (-1 unit) for an X-chromosome
encoded gene
(mutant 1). The potential contribution to the net aggregation pressure thus falls from 20
to 19 units. This decrease in aggregation pressure might be corrected in female zygotes in
one step if a second gamete happened to have a perfectly matching concentration-excess
mutant (+1 unit). The figure shows correction in two steps.
The partially compensated
decrease in aggregation pressure (39.5 units) in the organism formed by mutants
1 and 2,
is tolerated for a sufficient number of generations for a second concentration excess
mutant to arise (mutant 3) to finally rebalance the genome. However, the relative balance
between X-chromosome encoded products and autosomally encoded products would have been
changed. A back-cross with the original wild type would generate females with normal
aggregation pressures, but, owing to hypertranscription of the solitary X-chromosome in
the heterogametic sex, there would be an imbalance in males. |
|
The idea that Haldane's rule reflects an
X-chromosome/autosome incompatibility is an old one (Dobzhansky,
1937). However, studies
of hybrids between different fruit fly species have been interpreted by Coyne (1985) as
supporting an alternative X-chromosome/ Y-chromosome incompatibility theory. Wu (1992)
noted that Coyne only considered hybrid sterility.
Recently, Orr (1993) provided evidence that hybrid non-viability
reflects an X-chromosome/autosome incompatibility, in keeping with the arguments
presented here.
The founders of the "modern
synthesis" proposed that speciation required the divergence of many "ordinary" genes. Thus, speciation is a collective gene
function (Dobzhansky, 1937; Muller,
1942). Although there are some special cases, in
general, subsequent work has supported the hypothesis (Coyne,
1992). For example, at least
150 genes are implicated in the inviability of hybrids between various grasshopper races
which are believed to have arisen quite recently (Barton & Hewitt,
1981). The role of
a possible "speciation gene" involved in hybrid
male sterility in fruit fly (Orr, 1992), remains to be determined.
I present evidence elsewhere that fine-tuning
occurring at the DNA level ("GC/AT pressure"), may
also contribute to speciation (Forsdyke,
1994c).
4.5. RECOMBINATION HELPS GENES PREDICT THEIR FUTURE ENVIRONMENT
A major theme of this paper is that alleles compete
to get into the next generation, not only on the basis of differences in the specific
functions that they encode, but on the basis of their abilities to be "good mixers" and fit in with other genes in contributing to
collective functions, particularly the function concerned with generating a cytosolic
aggregation pressure. Recombination cross-over between genes serves this purpose in that
it separates emerging gene coalitions, thus effectively homogenizing the genome and
ensuring that each allele "knows" with some
certainty what aggregation pressure its products can expect to encounter in the next
generation (Dawkins, 1990).
4.6.
FINE-TUNING IN ASEXUAL SPECIES
In both sexual and asexual species an ability to
aggregate specifically proteins encoded by an intracellular pathogen would appear
advantageous. The effectiveness of this would depend on the effectiveness of the
fine-tuning of intracellular protein concentrations. Since the pressure to aggregate would
be greatly affected by ambient temperature (Lauffer,
1975), homeothermic species would be
able to fine-tune more precisely than poikilothermic species. Homeothermy is encountered
in sexual species, but many sexual species are also poikilothermic.
In sexual species, because of the "horizontal" mingling of genomes, mutations affecting the
concentration of a protein can be compensated by other mutations and can spread (Fig. 3).
In asexual species, a genomic response to an adverse mutation requires a transmission
"vertically" through the generations until
compensating mutations can occur (Muller,
1932). Thus, fine-tuning in asexual organisms
might depend more on the direct elimination of concentration mutants, rather than on the
emergence of compensating mutants.
|
This work was supported by the Leukaemia Research Fund
(Toronto), the Canadian Medical Research Council and the School of Graduate Studies of
Queen's University. I thank Mr. H. Metz for assistance with the figures.
REFERENCES
BAKER, B. S. & BELOTE, J. M. (1983). Sex determination and
dosage compensation in Drosophila melanogaster. A. Rev.
Genet. 17, 345-393.
BARTON, N. H. & HEWITT, G. M. (1981). The genetic basis of
hybrid inviability in the grasshopper Podisma pedestris. Heredity
47, 367-385.
BOOK, J. A. & SANTESSON, B. (1961). Nuclear sex in triploid
XXY human cells. Lancet, 2,
318.
BRIDGES, C. B. (1925). Sex in relation to chromosomes and genes.
Am. Nat. 59, 127-137.
BRIDGES, C. B. (1939). Cytological and genetic basis of sex. In:
Sex and Internal Secretions, 2nd
edn (Allen, E., ed.) pp. 15-63. Baltimore: Williams and Wilkins.
CHARLESWORTH, B. (1991). The evolution of sex chromosomes. Science 251, 1030-1033.
COLCLASURE, G. C. & PARKER, J. C. (1991). Cytosolic protein
concentration is the primary volume signal in dog red cells. J.
gen. Physiol. 98, 881-892.
COYNE, J. A. (1985). The genetic basis of Haldane's rule. Nature, Lond. 314, 736-738.
COYNE, J. A. (1992). Genetics and speciation. Nature, Lond. 355, 511-515.
DAWKINS, R. (1990). Parasites, desiderata lists and the paradox
of the organism. Parasitol. 100,
S63-S73.
DOBZHANSKY, T. (1929). The influence of the quantity and quality
of chromosomal material on the size of cells in Drosophila melanogaster. Roux Arch. Entwickim. 115, 363-379.
DOBZHANSKY, T. (1930). Genetical and environmental factors
influencing the type of intersexes in Drosophila melanogaster. Am.
Nat
. 64, 261-271.
DOBZHANSKY, T. (1937). Genetic nature of species differences. Am. Nat. 71, 404-420.
DOBZHANSKY, T. & SCHULTZ, J. (1934). The distribution of sex
factors in the X-chromosome of Drosophila melanogaster. J.
Genet
. 28, 349-386.
DORER, D. R., CHRISTENSEN, A. C. & JOHNSON, D. H. (1990). A
novel RNA helicase gene tightly linked to the Triplolethal locus of Drosophila. Nucleic Acids Res. 18, 5489-5494.
FORSDYKE, D. R. (1985). Heat shock proteins defend against
intracellular pathogens: a non-immunological basis for self/not-self discrimination. J. theor. Biol. 115, 471-473.
FORSDYKE, D. R. (1991). Early evolution of MHC polymorphism. J. theor. Biol. 150, 451-456.
FORSDYKE, D. R. (1994a). The heat shock response and the
molecular basis of genetic dominance. J. theor. Biol.
167, 1-5.
FORSDYKE, D. R. (1994b). Relationship of X-chromosome dosage
compensation to intracellular self/not-self discrimination: a resolution of Muller's
paradox? J. theor. Biol. 167, 7-12.
FORSDYKE, D. R. (1994c). A stem-loop "kissing" model
for the initiation of recombination and the origin of introns. FASEB.
J. 81 A1395.
FORSDYKE, D. R. (1994d). Hierarchy of frequencies of
complementary trinucleotide pairs depends on percentage G + C: implications for
speciation. Proc. Can. Fed. Biol. Socs. 37,
152.
FORSDYKE, D. R. (1995). Entropy-driven protein self-aggregation
as the basis for inter-species discrimination in the crowded cytosol. J. biol. Systems 39, 273-287.
FULTON, A. B. (1982). How crowded is the cytoplasm? Cell 30, 345-347.
GORMAN, M., KURODA, M. I. & BAKER, B. S. (1993). Regulation
of the sex-specific binding of the maleless dosage compensation protein to the male
X-chromosome in Drosophila. Cell 72,
39-49.
HARNDEN, D. G. (1961). Nuclear sex in triploid XXY human cells. Lancet 2, 488.
HITCHCOCK, D. I. (1924). Proteins and the Donnan equilibrium. Physiol. Rev. 4, 505-531.
HODGKIN, J. (1990). Sex determination compared in Drosophila
and Caenorhabditis. Nature, Lond. 344,
721-728.
HURST, L. D. (1993). The incidences, mechanisms and evolution of
cytoplasmic sex-ratio distorters in animals. Biol. Rev.
68, 121-193.
JABLONKA, E. & LAMB, M. J. (1990). The evolution of
heteromorphic sex chromosomes. Biol. Rev.
65, 249-276.
JACOBS, P. A. & MIDGEON, B. R. (1989). Studies of
X-chromosome inactivation in trisomies. Cytogenet. Cell
Genet. 50, 75-77.
JAFFE, E. & LAIRD, C. (1986). Dosage compensation in Drosophila.
Trends Genet. 2, 316-321.
KEYES, L. N., CLINE, T. W. & SCHEDL, P. (1992). The primary
sex determination signal of Drosophila acts at the level of transcription. Cell 68, 933-943.
LAUFFER, M. A. (1975). Entropy-Driven
Processes in Biology. New York: Springer-Verlag.
LAUGE, G. (1980). Sex determination. In: The Genetics and Biology of Drosophila, Vol.
2d. (Ashburner, M. & Wright, T. R. F., eds) pp. 33-106. New York: Academic Press.
LINDSLEY, D. L., SANDLER, L., BAKER, B. S., CARPENTER A. T.,
DENELL, R. E., HALL, J. C., JACOBS, P. A., MIKLOS, G. L., DAVIS, B. K., GETHMAN R. C.,
HARDY, R. W., HESSLER, A., MILLER, S. M., NOZAWA, H., PARRY, D. M. & GOULD-SOMERO, M.
(1972). Segmental aneuploidy and the genetic gross structure of the Drosophila genome. Genetics 71, 157-184.
LUCCESI, J. C. & MANNING, J. E. (1987). Gene dosage compensation in Drosophila
melanogaster. Adv. Genet.