by Donald R. Forsdyke
Copyright Elsevier, which permits "scholarly posting" of accepted manuscripts "provided that these are not for purposes of commercial use or systematic distribution." This differs slightly from the final edited version.
![]()
![]()
Keywords: Base composition; Epstein-Barr virus; Immune evasion; Purine-loading; Retrovirus 1. IntroductionA recent report by Lawrie et al. [1] of genome-wide strong negative (purifying) selection at synonymous (non-amino acid- determining) sites in the fruit fly, marks the continuing, but slow, convergence of the ideas of population geneticists with those of biochemists and biophysicists. Prior to the emergence of nucleic acid sequencing technology in the late 1970s, the latter groups had, through studies of base composition, equilibrium gradient sedimentation and thermal denaturation profiles, demonstrated the segmentation of genomes into units of relatively uniform base composition (GC%), which might relate either to genes ("homostability regions" or "microisochores"), or to larger regions potentially encompassing many genes ("isochores" or "macroisochores") [2, 3]. Departing from the view of many population geneticists that the evolutionary pressure on synonymous sites was selectively neutral [4, 5], it was proposed that synonymous sites were functional and subject to selection. For example, in 1976 Japanese biophysicists suggested [6]: "It is quite plausible -- that the homostability region plays an important part somewhere in the biological process within which the DNA is closely related. If so, then the evolutionary selective force can be considered to have fixed such regions in DNA. From the size of the homostability region, recombination might be one possible process which is aided by it. In any case, the wobble bases [at synonymous sites] must give the necessary redundancy to make a homostability region without spoiling the biological meaning of the genetic code: the activity of proteins." Consistent with this, analyses of the first nucleic acid sequences to become available led Richard Grantham to his "genome hypothesis" [7]. Here he proposed the profound functional significance of the genome as a whole, rather than of the genes it carried through the generations. "We conclude that mRNA sequences contain other information than that necessary for coding proteins. This other 'genome-type' information is mainly in the degenerate bases of the sequence. Consequently, it is largely independent of the amino acids coded." That the information obtained from protein-encoding parts of genes was indeed likely to be of 'genome type' became evident when the GC% of sequences flanking genes was found to correlate closely with that of synonymous codon positions [8, 9]. In 1986 Grantham et al. [10] imagined a shortly impending convergence of neutralist and selectionist viewpoints: "Although codon use is a characteristic of the genotype, most evolutionary analyses have been based on the phenotype. How much independence exists between the two levels of evolution has not been determined, although neutralists and selectionists are converging, which should help to find a solution. Possibly, future data on the relative rates of silent and non-silent mutations will help to clarify this situation." However, Grantham, although suspecting it was nucleic acid structure that was conserved at synonymous sites, focussed only on mRNA structure. Indeed, decades later, while affirming that "the underlying biological function disrupted by these [synonymous] mutations is unknown, but it is not related to the forces generally believed to be -- shaping the evolution of synonymous sites," Lawrie et al. [1] still held that "a strong possibility remains that the function underlying the strong constraint at synonymous sites is related to mRNA structure." Likewise, in 2013 Lind and Andersson [11] concluded that "the deleterious effects of synonymous mutations are not generally due to codon usage effects, but that mRNA secondary structure, is a major fitness constraint," and Park et al. [12] claimed "a major role of natural selection at the mRNA level in constraining protein evolution." Of course, structure conservation at the mRNA level means that conservation is also at the DNA level of the corresponding genes. Which is primary and which is secondary? Since, the potential of duplex DNA to adopt stem-loop configurations is often better developed in non-genic regions and introns, than in exons [13-16], the pressure for conservation could have arisen primarily at the genome level (be the genome itself DNA or RNA) . Indeed, in 1986 Bernardi and Bernardi [8] referred to selection acting on the "genome phenotype" rather than on the conventional phenotype. This would be consistent with a selective pressure relating to recombination [6]. Mutations likely to affect recombination are of two general forms, those affecting the proteins that mediate recombination (conventional phenotype), and those affecting their targets - DNA itself (genome phenotype). The former mutations would be localized to genes (mainly non-synonymous mutations), the latter would be genome-wide (synonymous and non-synonymous when within protein-encoding regions), and could involve DNA structure. The forces manifest when such mutations occur, should also operate in organisms with RNA genomes. Because of high mutation rates and high clinical interest, studies of retroviral evolution now provide well-documented exemplars of the general forces operating on genome sequences [17]. These forces can either promote or decrease recombination. In the latter case, new species can arise. While mechanisms of speciation are controversial, there is general agreement that "mutation is crucial in speciation because reproductive barriers cannot be generated without mutations" [18]. But, for RNA viruses in general, Simon-Loriere and Holmes [19] have argued that "there is little evidence that recombination is favoured by natural selection to create advantageous genotypes or purge deleterious mutations, as predicted if recombination functions as a form of sexual reproduction." However, as reviewed here, at least in the case of HIV-1, studies of RNA structure and base composition suggest otherwise. 2. Recombination, rejuvenation and conservation of RNA structureAt its most fundamental level, sex is recombination [20, 21]. The conjugation required for the meeting of paternal and maternal human gametes is an elaborate preliminary to the final meiotic meeting of parental genomes in the gonads of their offspring, where recombination occurs. The early microscopists who witnessed this final meeting described it as a "conjugation of the chromosomes" that was necessary for a rejuvenating "interchange of substances" "The conjugation of the chromosomes in the synapsis stage may be considered the final step in the process of conjugation of the [parental] germ cells. It is a process that effects the rejuvenation of the chromosomes; such rejuvenation could not be produced unless chromosomes of different parentage joined together, and there would be no apparent reason for chromosomes of like parentage to unite." In contrast to humans, retroviral sex is quite
elementary [23]. Yet, like humans, retroviruses are diploid. This diploidy in
HIV-1 can be heterozygous due to the viral strategy of mutation to near
oblivion, so countering host defences. The degree of heterozygosity in two HIV
genomes that are copackaged within an infectious retrovirus particle, if below
the sequence difference threshold above which recombination is inhibited (see
later), will allow viral rescue by recombination. Thus, within the next host
cell, the two partially crippled genomes can repeatedly recombine to generate a
rejuvenated form that will successfully colonialize the vulnerable population of
host cells (usually T4 lymphocytes) [24]. This implies that the virus will have
accepted mutations supporting the ability to recombine. Indeed, if RNA structure
were required for recombination, then HIV-1 genomes might display
higher conservation of RNA structure than of the encoded proteins.
Evidence for this postulate was presented in 1995 [25] and is now well supported
[26-30]. Furthermore, beyond its recognized regulatory roles, viral RNA
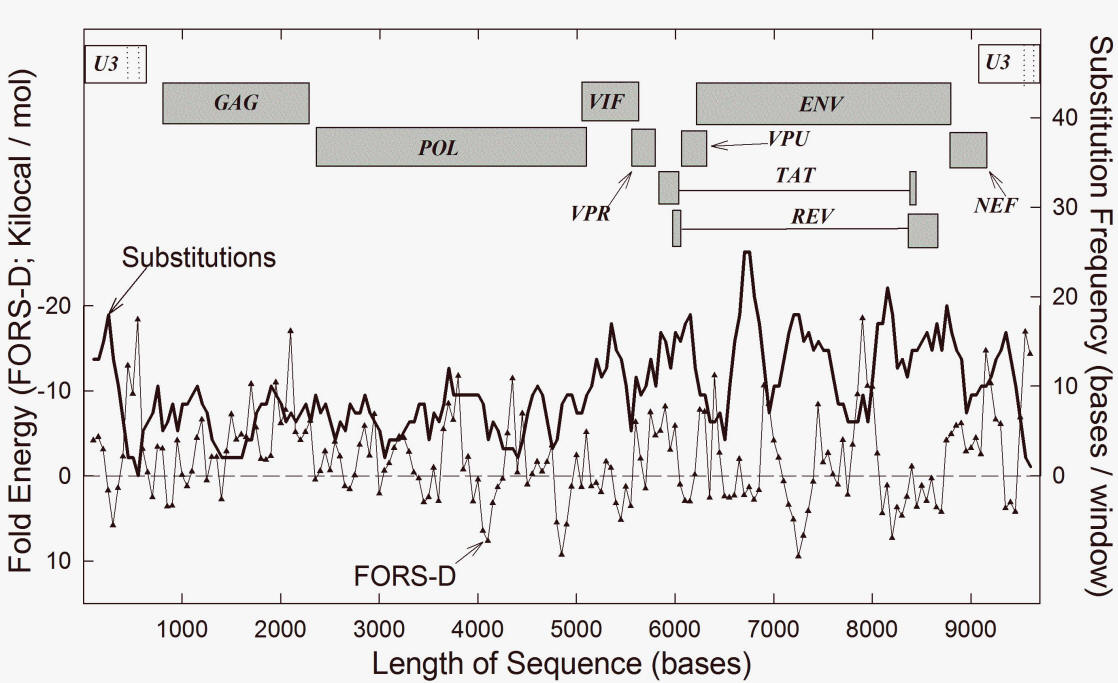
structure can be related to recombination and speciation [15, 16, 25]. Apart from this, HIV-1 genomes have high and stable levels of the purine A [31]. This A-richness has been related to the phenomenon of purine-loading and has implications for RNA structure [32, 33]. In keeping with current conventions [34], "high A%" here implies a high frequency of the base A. "High AT%" implies a high frequency of the combination of bases A and T, without specification of which of the two bases makes the major contribution. The same considerations apply to high values for GC% and AG% ("purine loading"). 3. Purine-loadingThe A-bias in HIV-1 is a characteristic of coding regions, but not of the non-coding long terminal repeats (LTRs). This parallels the selective "purine-loading" (AG% enrichment) of the encoding (mRNA synonymous) strands of the exons of genes of most biological species. Since purines do not pair with purines, this would militate against host self RNA-self RNA interactions. A purine-loaded virus would be less likely to form base-paired RNA duplexes with the host RNAs that appear when the genomes of stressed cells are pervasively transcribed (i.e. this would prevent self RNA-not-self RNA interactions) [34]. Preventing formation of double-stranded RNA (dsRNA) should be advantageous to the virus, since segments of dsRNA can provide alarm signals to the host, so alerting immunological defences [35]. Purine-loading also provides a rationale for interruption of proteins by low complexity segments corresponding to purine-rich codons [36, 37]. This implies function at the nucleic acid level. In other words, removing such segments from proteins should not affect their function. Thus, in 2011 Muralidharan et al. [38], from studies of a P. falciparum protein with an asparagine repeat sequence (corresponding to purine-rich codons GAY), concluded that "the asparagine repeat is dispensible for protein expression, stability and function." A similar observation has been made for a repeat within the purine-rich mRNA encoding the EBNA-1 protein, which normally maintains the Epstein-Barr virus (EBV) in a deeply latent state with its many pyrimidine-rich mRNAs unexpressed. In this way EBV should evade host immune defences [35, 39, 40]. Irrespective of whether avoidance of dsRNA is the correct adaptive explanation, purine loading is a fact. A-loading by HIV-1 could be an extreme expression of this [35]. Purine-loading can influence amino acid composition. Since the extra purines generally locate to third codon positions (the main site for synonymous mutations), it is likely that purine-loading has driven amino acid composition, rather than the converse [41]. The purines would be expected to occupy, and are indeed found in, the loop regions of the highly structured HIV genome [26, 33]. Indeed, in 2013 Mayrose et al. [42] noted pervasive synonymous site conservation in HIV-1, and Zanini and Neher [43] found that most synonymous mutations disrupt base pairing in HIV RNA stems and suggested direct fitness effects of the stem-loop structures. Values for AG% and GC% are reciprocally related, and purine-loading is achieved mainly by replacing C with A, leaving T and G relatively constant (so avoiding runs of Ts and Gs that can be detrimental). Thus, there can be conflict between 'AG pressure' and 'GC-pressure' [34, 41]. To appreciate why HIV-1 is A-loaded, not G-loaded, we return to the role of GC%. 4. Two species in one cell differ greatly in GC%Consider three examples of pairs of viral species, where members of each pair might occupy the same host cell. (1) An early report on species differences in GC% [44] noted that, while there was no clear relationship between the GC% values of virus and host, two insect viruses (polyhedral and capsule) with a common larval host (spruce budworm) differed dramatically in their GC% values (38% versus 51%). In this example it was uncertain whether these viruses could have shared a common cell within their larval host. However, in 1991 Schachtel et al. [45] noted for humans, that two neurotropic alphaherpesviruses also differ dramatically in GC% (46% versus 68%). The differences were dispersed, being present in both non-coding and coding regions and, in the latter case, mainly affected the third bases of codons (consistent with the close similarity in amino acid sequences of the viral proteins). But, as in the case of purine-loading (see above), the differences were sometimes sufficient to change amino acids (e.g. arginine to lysine). It was proposed that the differences were adaptive in that there would be less competition for host resources: "More specifically, it is proposed that HSV1 and VSV avoid competition for host resources such as nucleic acid precursors and aminoacyl-tRNAs through divergent base composition and codon usage. This may enable these two closely related viruses to coexist in the same host species and even to multiply simultaneously in the same cells." In 1994 a similar explanation was advanced by Bronson and Anderson [46] who contrasted the AU-richness of HIV-1 (42% GC), with the GC-richness of HTLV (53% GC). They supposed that the intracellular environment would consist of a variety of metabolic "ecological niches." Two viruses that coinfected the same host cell would avoid competition with each other by making different metabolic demands on NTP pools. There is, however, another interpretation. 5. Coinfectants either blend or speciateThe wide GC% differences between coinfecting viruses can be seen as part of the speciation process [15, 16, 25]. Two species of virus derived from a common ancestral species, which coexist synchronously within the same host cell (sympatry), have the opportunity to recombine. Due to this 'blending inheritance,' their reproductive isolation would then be lost and they would mutually destroy each other as independent species. To the extent that retaining their differentiation as independent species is advantageous, there would have been a strong selective pressure on their nucleic acids (i.e. on their genome phenotypes) to evolve to prevent recombination. If differences in GC% could achieve this (see later), then they would have been mutually driven to different GC% poles. If HIV-1 were the first to A-load (i.e. accept mutations to A), then it would have been driven to the low GC% pole. HTLV would have been driven to the high GC% pole. Since, to arrive at high GC% values, organisms preferentially replace A with C [41], HTLV would have foregone the advantages of purine-loading and adopted an alternative evolutionary strategy [35]. With its preference for A, HIV-1 recombinationally drove HTLV-1 away. We should note that, like the GC-rich EBV (see above), HTLV-1 is deeply latent and many carriers are symptom-free. Also like EBV, the duplex HTLV-1 provirus avoids transcribing its many pyrimidine-rich RNAs (controlled by the 5' LTR region). This is achieved by expressing a protein (HBZ) with a role analogous to that of the EBNA-1 protein (the latter encoded by a purine-rich mRNA). HBZ mRNA is transcribed from the antisense DNA strand as a purine-rich RNA species (controlled by the 3' LTR) [47]. 6. FORS-D analysis and SHAPE illuminate RNA structureNew technologies have greatly assisted the understanding of HIV RNA structure. In 1988 Le et al. [48, 49] used novel energy-minimization folding algorithms to show that RNA regions involved in intra-molecular base pairing tend to be more evolutionary stable. This stability of nucleic acid folding into stem-loop conformations is expected to be influenced by base composition, being low in low GC% organisms such as HIV-1 (since there is less opportunity for base-stacking with pairing between Gs and Cs). However, GC% values tend to characterize whole genomes or large genome sectors (see above). Base order is a character that critically affects local structure. A method of dissecting out the base order-dependent component of the folding energy - "folding of randomized sequence difference" (FORS-D) analysis (for details see [50]) - revealed many evolutionarily conserved structural elements in the HIV RNA genome; these were separated by less structured variable regions likely to be under positive Darwinian selection [25, 29, 42, 43]. These observations were recently confirmed and extended by "selective 2'-hydroxyl acylation analysis by primer extension" (SHAPE) [26]. While certain conserved structures corresponded to established regulatory elements, others corresponded to the peptide linkers between the protein domains of polyproteins. The results were interpreted in terms of "another level of genetic code" so that "higher ordered RNA structure directly encodes protein structure." Recombination was not mentioned. Others agreed, stating that "this novel component of the genetic code represents the strongest determinant of conservation" [29]. Again, there was no suggestion of a relationship to recombination. 7. Recombination depends on structure and speciesIf the degree of heterozygosity of two HIV-1 genomes, copackaged within an infectious retrovirus particle, is insufficient to generate sequence differences of a magnitude that inhibits recombination and fosters speciation (see below), then, by virtue of similarities in sequence and structure, the two genomes can generate recombinants with properties more advantageous than those of the originating parental genomes. But HIV-1 itself, by virtue of its high mutation rate, explores sequence space in years, to an extent that would take its host millions of years. Thus, as sequence differences increase, a differentiation of HIV-1 into "quasispecies" (up to approx. 15% intrasubtype variation), "subtypes" (approx. 15% - 30% intersubtype variation) and "groups" (>30% intergroup variation), is recognized. How efficiently can recombination have checked this variation? Can differences in recombination between members of these various categories, although mechanistically copy-choice (template switching without strand breakage), guide our understanding of speciation in more complex organisms where recombination is likely to involve DNA strand breakage [15, 16]? Recombination in HIV-1 begins with the formation of RNA dimers by means of complementary loop-loop "kissing" interactions between "dimer initiation sequences" (DIS) that are part of stem-loop secondary structures in the Ψ (packaging signal) region of the genome. A similar DNA "kissing" may be involved in recombination in higher organisms [15, 51]. A lack of base complementarity between HIV-1 DIS loops decreases recombination, but does not eliminate it. This suggests that other parts of the genome can assist dimer formation. Upon transfer to a new host cell, copy choice recombination occurs between the colocalized genomes in HIV-1 homodimers or heterodimers [52, 53]. Since a species is defined by its recombinational (reproductive) isolation relative to other species [16, 18], it is important to note that, albeit rare, recombination can occur between members of different HIV-1 groups [54]. This defines them as belonging to the same species. On the other hand, although HIV-1 and HIV-2 can coinfect an individual, no recombinants have been detected, so they are different species. HIV-1 intrasubtype recombination is more frequent than intersubtype (intragroup) recombination, which is more frequent than intergroup recombination [55]. Thus, after introduction of synonymous substitutions, it was found that 5%, 9%, and 18% sequence differences corresponded, respectively, to 35%, 74% and 95% decreases in recombination frequency [52]. Others, with a different assay, found 9% and 18% sequence differences corresponded to 67% and 90% decreases in recombination [53]. These values are consistent with previous FORS-D studies (see below), which postulated that HIV-1 secondary structure is conserved because, in addition to regulatory roles, it plays a critical role in recombination [25]. Consistent with this, in 2011 Simon-Loriere et al. noted [28]: "Strong disparities [in recombination] were observed for comparable degrees of sequence identity in conserved regions, indicating that -- parameters other than the level of sequence identity modulate -- recombination. -- [R]egions of the genomic RNA with a high proportion of residues involved in the formation of secondary structure contained significantly more [recombinational] breakpoints. The extent of RNA structure along the HIV genome seems to provide us with a relatively accurate picture of the pattern of recombinant genomes generated by the mechanism of recombination." 8. Base order conserves structureAs the differences between two subtypes increase, it becomes evident that conservation (low substitution rate) corresponds to regions where bases are ordered to support the formation of higher ordered local structure (i.e. the base order-dependent component of stem-loop potential is maximized). Thus, when fold energy is high, substitutions are low. This is shown in Fig. 1 (taken from ref. 25) where subtype HIVSF2 differs by 455 dispersed substitutions (4.68% difference) from the reference sequence (HIVHXB2). Here structural stability in 200 base moving windows (expressed in negative kcal/mol), is plotted with the corresponding substitution frequency for each window. In general, where the base order-dependent folding stability is high, substitutions are low. Where base order-dependent folding stability is low, substitutions are high. In other words, base conservation associates with stable RNA structure.
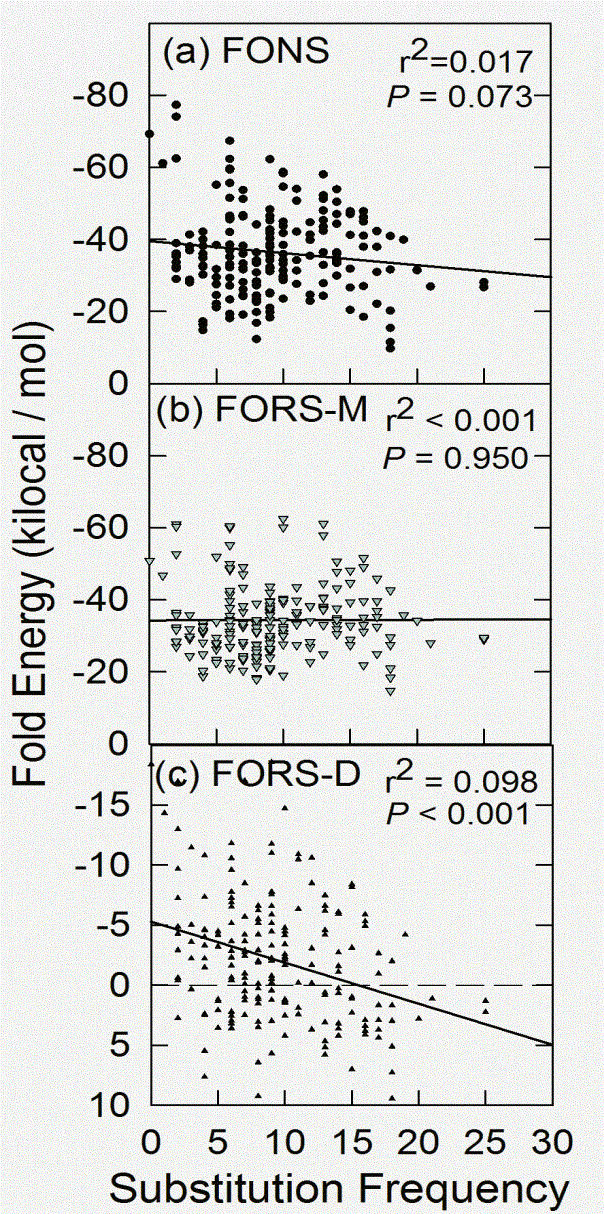
This relationship is better seen when the two values within each window are plotted together. The reciprocal relationship does not hold for the base composition-dependent component (Fig. 2b), but does hold for the base order-dependent component (Fig. 2c). Although the points are widely scattered, approximately 10% of the decline in the base order-dependent component of the folding energy is accounted for by increasing substitutions (r2 = 0.098).
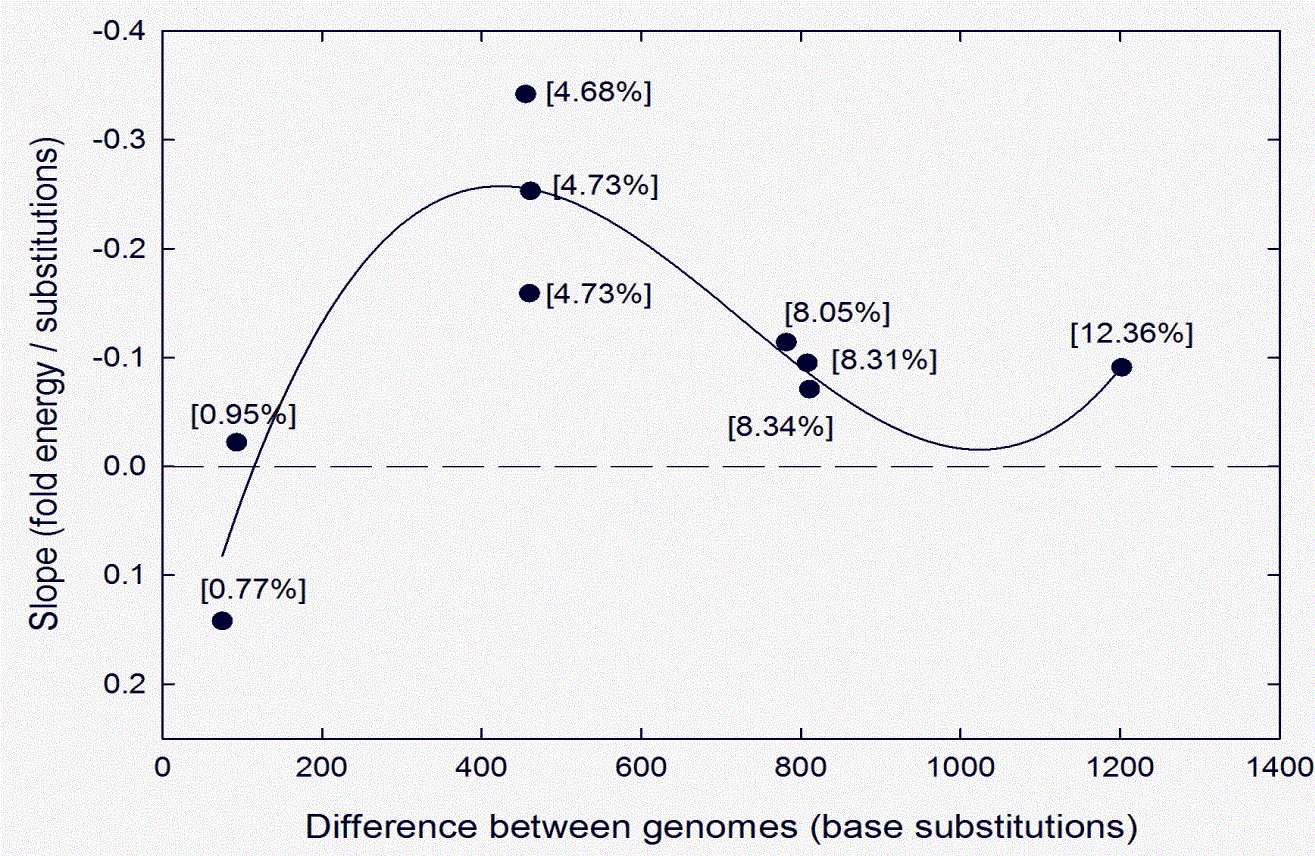
This result, together with the results of studies with other HIV-1 pairs whose sequences differed more or less than in this case, are summarized in Fig. 3 (adapted from Table 1 of Ref. [25]). When differences between HIV-1 genomes are low (e.g. 0.77%; 75 substitutions) a correlation between stable structure and conservation is difficult to demonstrate (slope value 0.142, which is not significantly different from zero). However, when differences are intermediate (4.68%; 455 substitutions; see Figs. 1 and 2) the correlation is significant (slope value -0.34; P < 0.001). Thus, in a sequence window where bases are conserved (low substitutions), the bases are likely to contribute positively, by virtue of their ordering, to RNA structure. Conversely, in a sequence window where base order is variable (high substitutions) the bases do not contribute to RNA structure (or can sometimes contribute negatively; i.e. positive FORS-D values).
With higher differences between genomes (8%, 12%), slope values decline and are of marginal significance. This implies that the additional substitutions are now entering windows corresponding to conserved regions, where the substitutions can change both base order and composition, so modify RNA structures that, when substitutions were less, would have supported intra-species recombination. Thus, there is a difference threshold above which recombination, with its associated rejuvenating effects (see above), begins to fail. 9. Base composition and phylogenetic analysisAbove the threshold the potential to follow a new evolutionary path - speciation potential - increases. In this circumstance base composition plays a more crucial role. A very small fluctuation in GC% is able to substantially change structure so that 'kissing' interactions fail and speciation can initiate [20, 56]. Once species are established, in phenotypically more complex organisms GC% values may then turn to other roles, but in viruses that have not undergone extensive phenotypic adaptation this seems not to occur, and 'echoes' of the originating GC% differences may remain (see above) [15, 44, 45]. Such differences have been found
useful in the phylogenetic analyses of retroviruses [46]. Likewise, for
influenza viruses Sampath et al. [57]
reported that different "evolving virus species" can be differentiated on the
basis of GC% differences: "Base composition derived clusters inferred from this
[phylogenetic] analysis showed 100% concordance to previously established
clades." Analyses of influenza virus RNA structures show that, as in the case of
HIV-1, conserved RNA structure is in potential conflict with other functions.
For example, Moss et al. [58] found
for influenza virus that "RNA structural constraints lead to suppression of
variation in the third (wobble) position of amino acid codons." While GC% differences usually do not suffice for phylogenetic analysis in more complex organisms, palindrome frequencies can be informative [59]. Higher ordered nucleic acid structure depends on appropriately placed, complete or incomplete, palindrome-like inverted repeats of distinctive base order and composition, consistent with Chargaff's second parity rule [32]. The frequency of short palindromes has low intra-species variance, but high inter-species variance. Mutations sufficient to generate the large variances are sometimes cryptic, in that there are no obvious phenotype differences [60]. Yet, such palindrome frequencies can be used to distinguish species. 10. Gene definition and recombinationIt is suggested that the potential to adopt a higher order genome structure relates to recombination [25, 27, 28]. Conserved structure may influence where initial strand-switching occurs when there is copy-choice recombination, and where initial crossovers occur in conventional recombination [15, 24]. Recombination is also linked to the seemingly endless debate on how to define a gene [61]. There is an apparent discrepancy between the gene as defined by biochemists and the gene defined by G. C. Williams and R. C. Dawkins as a "selfish" element that is able to resist recombinational disruption. Evidence that final recombinational crossovers are preferentially located close to gene boundaries brings the two definitions into close correspondence [62]. Studies of the HIV-1 genome further support this. In 1999, for intergroup recombination, Takehisa et al. [54] showed that: "Breakpoints appeared mostly near the boundaries of the respective genes. The high frequency of recombination that occurs only near the beginning or end of the respective genes seems to reflect a common adaptive strategy for recombination." As noted above, SHAPE analysis [26] indicates that conserved structure corresponds to the interdomain regions of various polyproteins. Further application of SHAPE led Simon-Loriere et al. [27] to conclude that: "Junctions between genes are enriched in structured RNA elements and are also preferred sites for generating functional recombinant forms. These data suggest that RNA structure-mediated recombination allows the virus to exchange intact genes rather than arbitrary subgene fragments, which is likely to increase the overall viability and replication success of the recombinant HIV progeny." Smyth et al. [24] agreed: "As junctions between genes are enriched with RNA structure -- one could argue that the HIV genome has evolved to exchange intact genes as genetic units, rather than as random fragments of the genome, which should increase the chances of recreating a viable virus." 11. Conclusions
AcknowledgementsMy AIDS studies were supported in the 1980s and 1990s by the American Foundation for AIDS Research and the Medical Research Council of Canada. Queen's University hosts my evolution education webpages where some of the cited references may be found. References[1] D.S. Lawrie, P.W. Messer, R. Hershberg, D.A. Petrov, Strong purifying selection at synonymous sites in D. melanogaster, PLOS Genetics 9 (2013) e1003527. [2] D.R. Forsdyke, Regions of relative GC% uniformity are recombinational isolators, J. Biol. Sys.12 (2004) 261-271. [3] S.-J. Lee, J.R. Mortimer, D.R. Forsdyke, Genomic conflict settled in favour of the species rather than of the gene at extreme GC% values, App. Bioinf.3 (2004) 219-228. [4] M. Kimura, Evolutionary rate at the molecular level, Nature 217 (1968) 624. [5] J.I. King, T.H. Jukes, Non-Darwinian evolution, Science 164 (1969) 788-798. [6] A. Wada, H. Tachibana, O. Gotoh, M. Takanami, Long range homogeneity of physical stability in double-stranded DNA, Nature 263 (1976) 439-440. [7] R. Grantham, Workings of the genetic code, Trends Biochem. Sci. 5 (1980) 327-331. [8] G. Bernardi, G. Bernardi, Compositional constraints and genome evolution, J. Mol. Evol. 24 (1986) 1-11. [9] O. K. Clay, G. Bernardi. GC3 of genes can be used as a proxy for isochore base composition: a reply to Elhaik et al., Mol. Biol. Evol. 28 (2011) 21-23. [10] R. Grantham, P. Perrin, D. Mouchiroud, Patterns in codon usage of different kinds of species, Oxford Surv. Evol. Biol.3 (1986)48-81. [11] P.A. Lind, D.I. Andersson, Fitness costs of synonymous mutations in the rpsT gene can be compensated by restoring mRNA base pairing, PLOS One 8 (2013) e63373. [12] C. Park, X. Chen, J.-R. Yang, J. Zhang, Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly, Proc. Natl. Acad. Sci. USA 110 (2013) E678-E686. [13] D.R. Forsdyke, A stem-loop "kissing" model for the initiation of recombination and the origin of introns, Mol. Biol. Evol 12 (1995) 949-958. [14] D.R. Forsdyke, Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes: an application of FORS-D analysis, Mol. Biol. Evol 12 (1995) 1157-1165. [15] D.R. Forsdyke, Different biological species 'broadcast' their DNAs at different (G + C)% 'wavelengths,' J. Theor. Biol. 178 (1996) 405-417. [16] D.R. Forsdyke, The origin of species revisited. A Victorian who anticipated modern developments in Darwin's theory, McGill-Queen's University Press, Montreal (2001). [17] R.A. Neher, T. Leitner, Recombination rate and selection strength in HIV intrapatient evolution, PLOS Comput. Biol. 6 (2010) e1000660. [18] M. Nei,
M. Nozawa, Roles of mutation and selection in speciation: from Hugo de Vries to
the modern genomic era, Genome Biol. Evol. 3 (2011) 812-829. [19] E.Simon-Loriere, E.C. Holmes, Why do RNA viruses recombine? Nat. Rev. Microbiol. 9 (2011) 617-626. [20] D.R. Forsdyke, Molecular sex: the importance of base composition rather than homology when nucleic acids hybridize,J. Theor. Biol. 249 (2007) 325-330. [21] R.E. Michod, H. Bernstein, A.M. Nedelcu, Adaptive value of sex in microbial pathogens, Inf. Genet. Evol. 8 (2008) 267-285. [22] T.J. Montgomery, A study of the chromosomes of the germ cells of metazoa, Trans. Am. Phil. Soc. 20 (1901) 154-236. [23] H.M. Temin, Sex and recombination in
retroviruses, Trends Genet. 7 (1991) 71-74. [24] R.P.
Smythe, M.P. Davenport, J. Mak, The origin of genetic diversity in HIV-1, Virus
Res. 169 (2012) 415-429. [25] D.R. Forsdyke, Reciprocal relationship between stem-loop potential and substitution density in retroviral quasispecies under positive Darwinian selection, J. Mol. Evol. 41 (1995) 1022-1037. [26] J.M. Watts, K.K. Dang, R.J. Gorelick, C.W. Leonard, J.W. Bess, R. Swanstrom, C.L. Burch, K.M. Weeks, Architecture and secondary structure of an entire HIV-1 genome, Nature 460 (2009) 711-716. [27] E. Simon-Loriere, D.P. Martin, K.M. Weeks, M. Negroni, RNA structures facilitate recombination-mediated gene swapping in HIV-1, J. Virol. 84 (2010) 12675-12682. [28] E. Simon-Loriere, P. Rossolillo, M. Negroni, RNA structure, genomic organization and selection of recombinant HIV, RNA Biol. 8 (2011) 280-286. [29] J. Snoeck, J. Fellay, I. Bartha, D.C. Douek, A. Telenti, Mapping of positive selection sites in the HIV-1 genome in the context of RNA and protein structural constraints, Retrovirol. 8 (2011) 87. [30] R. Sanjuan, A.V. Borderia, Interplay between RNA structure and protein evolution in HIV-1, Mol. Biol. Evol. 28 (2011) 1333-1338. [31] J. Kypr, J. Mrazek, Unusual codon usage in HIV, Nature 327 (1987) 20. [32] D.R. Forsdyke, J.R. Mortimer, Chargaff's legacy, Gene 261 (2000) 127-137. [33] D.R. Forsdyke, S.J. Bell, Purine-loading, stem-loops, and Chargaff's second parity rule: a discussion of the application of elementary principles to early chemical observations, Appl. Bioinf. 3 (2004) 3-8. [34] D.R. Forsdyke, Evolutionary bioinformatics, 2nd edition, Springer, New York, 2011. [35] A.D. Cristillo, J.R. Mortimer, I.H. Barrette, T.P. Lillicrap, D.R. Forsdyke, Double-stranded RNA as a not-self alarm signal: to evade, most viruses purine-load their RNAs, but some (HTLV-1, Epstein-Barr) pyrimidine-load, J. Theor. Biol 208 (2001) 475-491. [36] H.Y. Xue, D.R. Forsdyke, Low complexity segments in Plasmodium falciparum proteins are primarily nucleic acid level adaptations, Mol. Biochem. Parasitol 128 (2003) 21-32. [37] X. Tian, J.E. Strassmann, D.C. Queller, Genome nucleotide composition shapes variation in simple sequence repeats, Mol. Biol. Evol. 28 (2011) 899-909. [38] V. Muralidharan, A. Oksman, M. Iwamoto, T.J. Wandless, D.E. Goldberg, Asparagine repeat function in Plasmodium falciparum protein assessed via a regulatable fluorescent affinity tag, Proc. Natl. Acad. Sci. USA 108 (2011) 4411-4416. [39] S.R. Starck, S. Cardinaud, N. Shastri, Immune surveillance obstructed by viral mRNA, Proc. Natl. Acad. Sci USA 105 (2008) 9135-9136 [40] J.T. Tellam, L. Lekieffre, J. Zhong, D.J. Lynn, R. Khanna, Messenger RNA sequence rather than protein sequence determines the level of self-synthesis and antigen presentation of the EBV-encoded antigen, EBNA1, PLOS Pathog. 8 (2012) e1003112. [41] J.R. Mortimer, D.R. Forsdyke, Comparison of responses by bacteriophage and bacteria to pressures on the base composition of open reading frames, Appl. Bioinf. 2 (2003) 47-62. [42] I. Mayrose, A. Stern, E.O. Burdelova, Y. Sabo, N. Laham-Karam, R. Zamostiano, E. Bacharach, T. Pupko, Synonymous site conservation in the HIV-1 genome, BMC Evol. Biol. 13 (2013) 164. [43] F. Zanini, R.A. Neher, Quantifying selection against synonymous mutations in HIV-1, env evolution, J. Virol. 87 (2013) 11843-11850 doi:10.1128/JVI.01529-13 [44] G.R. Wyatt, The nucleic acids of some insect viruses, J. Gen. Physiol. 36 (1952) 201-205. [45] G.A. Schachtel, P. Bucher, E.S. Mocarski, B.E. Blaisdell, S. Karlin, Evidence for selective evolution on codon usage in conserved amino acid segments of alphaherpesvirus proteins, J. Mol. Evol. 33 (1991) 483-494. [46] E.C. Bronson, J.N. Anderson, Nucleotide composition as a driving force in the evolution of retroviruses, J. Mol. Evol. 38 (1994) 506-532. [47] L.B. Cook, M. Elemans, A.G. Rowan, B. Asquith, HTLV-1: persistence and pathogenesis, Virol. 435 (2012) 131-140. [48] S.-Y. Le, J.H. Chen, M.J. Braun, M.A. Gonda, J.V. Maizel, Stability of RNA stem-loop structure and distribution of non-random structure in human immunodeficiency virus (HIV-1), Nucleic Acids Res. 16 (1988) 5153-5168. [49] S.-Y. Le, J.V. Maizel, A method for assessing the statistical significance of RNA folding, J. Theor. Biol 138 (1989) 495-510. [50] D.R. Forsdyke, Introns first, Biol. Theor. 7 (2013) 196-203. [51] C. Danilowicz, C.H. Lee, K. Kim, K. Hatch, V.W. Coljee, N. Kleckner, M. Prentiss Single molecule detection of direct, homologous, DNA/DNA pairing, Proc. Natl. Acad. Sci. USA 106 (2009) 19824-19829. [52] A. Onafuwa-Naga, A. Telesnitsky, The remarkable frequency of human immunodeficiency virus type 1 genetic recombination, Microbiol. Mol. Biol. Rev. 73 (2009) 451-481. [53] O.A. Nikolaitchik, A. Galli, M.D. Moore, V.K. Pathak, W.-S. Hu, Multiple barriers to recombination between divergent HIV-1 variants revealed by a dual-marker recombination assay, J. Mol. Biol. 407 (2011) 521-531. [54] J. Takehisa, L. Zekeng, E. Ido, Y. Yamaguchi-Kabata, I. Mboudjeka, Y. Harada, T. Miura, L. Kaptue, M. Hayami, Human immunodeficiency virus type 1 intergroup (M/O) recombination in Cameroon, J. Virol. 73 (1999) 6810-6820. [55] M.P.S. Chin, J. Chen, O.A. Nikolaitchik, W.-S. Hu, Molecular determinants of HIV-1 intersubtype recombination potential, Virology 363 (2007) 437-446. [56] D.R. Forsdyke, An alternative way of thinking about stem-loops in DNA. A case study of the G0S2 gene, J. Theor. Biol 192 (1998) 489-504. [57] R. Sampath, K.L. Russell, C., Massire, M.W. Esho, V. Harpin, L.B. Blyn, R. Melton, C. Ivy, T. Pennella, F. Li, H. Levene, T.A. Hall, B. Libby, N. Fan, D.J. Walcott, R. Ranken, M. Pear, A. Schink, J. Gutierrez, J. Drader, D. Moore, D. Metzgar, L. Addington, R. Rothman, C.A. Gaydos, S. Yang, K. St. George, M.E. Fuschino, A.B. Dean, D.E. Stallknecht, G. Goekjian, S. Yingst, M. Monteville, M.D. Saad, C.A. Whitehouse, C. Baldwin, K.H. Rudnick, S.A. Hofstadler, S.M. Lemon, D.J. Ecker, Global surveillance of emerging influenza virus genotypes by mass spectrometry, PLOS One2 (2007) e489. [58] W.N. Moss, S.F. Priore, D.H. Turner, Identification of potential conserved RNA secondary structure throughout influenza A coding regions, RNA 17 (2011) 991-1011. [59] E. Lamprea-Burgunder, P. Ludin, P. Maser, Species-specific typing of DNA based on palindromic frequency patterns, DNA Res. 18 (2011) 117-124. [60] D.R. Forsdyke, Base composition, speciation, and barcoding, Trends Ecol. Evol. 28 (2013) 73-74. [61] D.R. Forsdyke, Scherrer and Josts' symposium. The gene concept in 2008, Theory Biosci. 128 (2009) 157-161. [62] D.R. Forsdyke, The selfish gene revisited: reconciliation of Williams-Dawkins and conventional definitions, Biol. Theory 5 (2011) 246-255.
Go to: Earlier arXiv Archive Version (Click Here) Return to: AIDS Page (Click Here) Return to: Bioinformatics Page (Click Here) Return to: Evolution Index Page (Click Here) Return to: HomePage (Click Here) This page was established Nov 2013 and was last edited on 11 November 2020 by Donald Forsdyke. |