Microsatellites scattered around genomes usually confer extrusion asymmetry on the region of DNA they occupy. Why this should occur is the subject of the following paper.
![]()
Microsatellites that violate
Chargaff's second parity rule have base order-dependent asymmetries in the
folding energies of complementary DNA strands and may not drive speciation
Journal of Theoretical Biology (2008) 254, 168-177
Copyright Elsevier Scientific
b The Clinical Experiment Center, the First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu 210029, China
c Department
of Biochemistry, Queen's University,
![]()
2. Crick's unpairing postulate
3. Chargaff's second parity rule and folding symmetry
4. Calculation of folding energies
5. Symmetrical distribution of folding energies of top and bottom strands
6. Asymmetrical distribution of folding energies in telomeres
7. Asymmetrical distribution of folding energies in internal STR regions
8. Asymmetrical distribution is greater with short repeat units
9. Asymmetrical distribution when second parity rule is violated
![]()
|
Abstract Models for meiotic recombination based on Crick's "unpairing postulate" require symmetrical extrusion of stem-loop structures from homologous DNA duplexes. The potential for such extrusion is abundant in many species and, for a given single-strand segment, can be quantitated as the "folding of natural sequence" (FONS) energy value. This, in turn, can be decomposed into base order-dependent and base composition-dependent components. The FONS values of top and bottom strands in most C. elegans segments are close, as are the corresponding base order-dependent and base composition-dependent components; any discrepancies are in the base composition-dependent component. This suggests that the strands would extrude with similar kinetics. However, interspersed among these segments and at the ends of chromosomes (telomeres) are segments containing short tandem repeats (microsatellites) which, by virtue of their high variability, have been postulated to inhibit the pairing of homologous chromosomes and hence drive speciation. In these segments there are usually wide discrepancies between the FONS values of top and bottom strands, mainly attributable to differences in base order-dependent components. Analyses of artificial microsatellites of different unit sizes and base compositions show that this asymmetrical distribution of folding potential is greatest for microsatellites when the units are short and violate Chargaff's second parity rule. It is proposed that when there is folding asymmetry, recombination proceeds by special, strand-biased, somatic mechanisms analogous to those operating with Chi sequences in E. coli. If meiotic recombination in the germ-line requires extrusion symmetry, then a general inhibitory influence of microsatellite-containing segments could mask the antirecombinational influence of their variability. Thus, microsatellites may not have driven speciation. |
Keywords: C.
elegans; Chi sequences; Crick's
unpairing postulate; Microsatellites; Recombination; Short tandem repeats;
Telomeres
|
Repetitive sequences form much of the genomes of organisms considered higher on the evolutionary scale and were postulated to play a role at the level of individual organisms (Britten and Davidson, 1971). However, the concept of "selfish DNA" led to the view that repetitive sequences might make "no specific contribution to the phenotype" (Orgel and Crick, 1980). Conceding this, it was argued that, since repetitive sequences tend to vary (a) more than unique sequences and (b) more between species than within a species, they "could act as a blunt instrument driving speciation by reproductive isolation alone, without reference to adaptation" (Robertson, 1981). Thus, repetitive sequences may be more important at the species, than at the individual, level. Sequence differences would impede the meiotic pairing of homologous chromosomes so impeding recombination and making interspecies hybrids infertile. Unable to continue the line through their hybrid, parents would be reproductively isolated from each other, and so would be deemed members of distinct species with the potential to pursue independent reproductive paths (Flavell, 1982). Consistent with this, it has been proposed that DNA sequence divergence ("chromosomal hypothesis"), rather than differences in gene function ("genic hypothesis"), underlies the majority of speciation events (Forsdyke, 2007a). We consider here whether divergence might be a general genomic property or might be driven by special non-genic sequences - namely, a class of highly variable repetitive sequence containing short tandem repeats (STRs) often referred to as "microsatellites" or "minisatellites" (Ellegren, 2004). |
2. Crick's unpairing postulate
|
Aggregations between molecules may be broadly classified as like-with-like and like-with-unlike. Like can aggregate with like by virtue of shared regularities in structure and/or resonance frequencies, as when pure crystals appear within a mixture of solutes in solution, or virus coat proteins aggregate (Lauffer, 1975; Muller, 1941). Like can aggregate with unlike by virtue of complementarity of structures and compatible resonance frequencies. Ostensibly, the pairing of homologous chromosomes in meiosis appears as an example of like-with-like aggregation. Yet, at the molecular level, Crowther (1922) drew an analogy with a sword pairing with its scabbard (i.e. complementarity of structure). Decades later the discovery of the double-helical structure of DNA revealed complementarity (A pairing with T, and G pairing with C) as a fundamental feature of this major chromosomal component (Watson and Crick, 1953). In his "unpairing postulate" Crick (1971) proposed that to initiate meiosis the strands in a DNA duplex would unpair thus allowing cross-pairing of strands without strand breakage (paranemic pairing). In other words, the "sword" of one duplex would pair with the matching "scabbard" of the other, and vice versa. If scabbards did not match (absence of homology) the pairing would fail, hence avoiding commitment to strand-breakage and launching a potential speciation event. Seeking broad generalizations, Crick considered the unpaired DNA strands as single-stranded bubbles. He did not explore the possibility that, by virtue of their palindrome content, the unpaired strands might rapidly adopt higher ordered stem-loop structures. In other words, the "swords" in single stranded sequences might find local "scabbards" within their own strands, thus perhaps militating against the cross-pairing between strands that he had postulated. Yet sequencing studies were already suggesting (since verified; e.g. (Forsdyke, 1995a) ) that the potential to assume local intra-strand stem-loop structures is abundant in nucleic acids of many species. Such structures usually contain sequences displaying a folding symmetry that can be described as "palindromic" since, at the duplex level, they read the same forward and backward. A duplex containing 5'AAAAAAAATTTTTTTT3' is considered palindromic, whereas a duplex containing 5'AAAAAAAACCCCCCCC3' is not (see below). A duplex with two complementary limbs separated by an intermediate region (e.g. 5'AAAAAAAAGGGGTTTTTTTT3') would form a stem and a loop (in this case G-rich in the top strand) and would be considered to display partial palindromicity. In 1974 Crick wrote:
While intra-strand pairing between complementary sequences that had adopted stem-loop configurations seemed to contradict Crick's model for inter-strand meiotic pairing, several more specific recombination models invoked intra-strand pairing, thus providing an adaptive explanation for the abundance of palindromes (Doyle, 1978; Sobell, 1972; Wagner and Radman, 1975). Consistent with the latter models, Tomizawa demonstrated for RNA that inter-strand pairing could follow exploratory "kissing" interactions between the loops in complementary single-strand molecules. Furthermore, Kleckner and Weiner (1993) suggested that the Tomizawa mechanism might apply to the pairing between stem loops extruded from the DNAs in homologous chromosomes (Zickler, 2006). Small differences in GC% (i.e. no violation of Chargaff's second parity rule; see section 3) would suffice to disrupt pairing and impede recombination (Forsdyke, 2006; 2007a). There is a well-known association of recombination with palindromes (Leach, 1994), and a role for stem-loop intermediates has been suggested from studies of recombination in HIV and in its coreceptor gene (Zhang et al., 2005a; 2005b). Recent studies have shown that, despite the presence of heterologous duplexes, homologous DNA duplexes can aggregate in simple salt solution in the absence of proteins. However, whether stem-loop intermediates are involved is undetermined (Baldwin et al., 2008). A prediction of the stem-loop models is that complementary kissing interactions would be favored if the "sword" strands and "scabbard" strands of a DNA duplex were extruded with similar kinetics (Forsdyke, 2006; 2007a). As an indicator of this possibility, the "top" ("W") strand of a duplex might show the same propensity to fold as its complementary "bottom" ("C") strand. There would then be symmetry between the folding potential of top and bottom strands. We here report studies with the nematode worm C. elegans (supported by our unpublished studies with other organisms) that show this is generally true. However, the symmetry is often lost in microsatellite-containing regions, raising the possibility that, by virtue of the asymmetry, microsatellites might exert a general antirecombinational effect. Yet, microsatellites are thought to promote, not impede, recombination (Ellegren, 2004). We explore here the basis of this apparent paradox. |
3. Chargaff's second parity rule and folding symmetry
|
For the nuclear
genomes of most biological species Chargaff's first parity rule for
duplex DNA (A = T, G = C) also applies, to a close approximation, to
single stranded DNA (Chargaff's second parity rule; Forsdyke
and Mortimer, 2000; Phillips et al., 1987; Prabhu, 1993; Rogerson,
1989). Thus, the AT-rich DNA sequence 5'AAAAAAAATTTTTTTT3'
obeys Chargaff's second parity rule and has the potential to fold
into a stem-loop (hairpin-like) configuration (Gierer,
1966). In an antiparallel duplex containing this sequence in its
"top" (W) strand (as listed in GenBank), the corresponding "bottom"
(C) strand, 3'TTTTTTTTAAAAAAAA5', has the same base composition and
the same potential to fold into a stem-loop configuration. Thus,
there is symmetry in the folding energies of top and bottom strands.
Furthermore, the two strands would display the same buoyant density
when subjected to centrifugation in alkaline CsCl gradients. In
contrast, the AC-rich sequence 5'AAAAAAAACCCCCCCC3' (top strand)
does not obey the second parity rule, has little potential to fold,
and has a different base composition from the corresponding GT-rich
bottom strand, 3'TTTTTTTTTGGGGGGGG5'. However, since G can form a
weak Watson-Crick base-pair with T (Allawi and
SantaLucia, 1998), the latter sequence retains some potential to
fold. Thus, there is asymmetry in the folding energies of top and
bottom strands. By virtue of their composition differences, the two
stands display different buoyant densities in alkaline CsCl
gradients. When banded in such gradients one strand can be regarded
as a "satellite" of the other. Indeed, a significant class of
repetitive sequences was discovered by virtue of this satellite
property (Flamm et al., 1969). Base composition and
order both influence folding symmetry. Thus, the degrees of
symmetry/asymmetry would be modified if the orders of the bases were
less simple than shown above (e.g. if each sequence were shuffled).
One strand of many
microsatellite sequences being GT-rich in violation of Chargaff's
second parity rule, the corresponding duplexes would be expected to
display folding asymmetry and hence impede meiotic pairing (see
Section 2). Yet, paradoxically, GT-richness appears to favor recombination.
In prokaryotes recombination may be associated with "GT-rich
islands" (approx. 1 kb) that contain strand-specific "crossover
hotspot instigator" sequences - Chi sequences (GCTGGTGG; E.
coli) or Chi-like sequences (GNTGGTGG; H.
influenzae; Bell et al., 1998; Lao and Forsdyke,
2000; Tracy et al., 1997). For their function Chi sequences require
a GT-rich base composition, but the order of bases is also
important. Specific GT-rich sequences (GGGGCTGGG) embedded in
GT-rich regions are also important in the strand-biased
recombination events that allow immunoglobulin class switching in
higher eukaryotes. Here folding asymmetry would facilitate access of
the mutagenic deoxycytosine deaminase to the AC-rich strand (Huang
et al., 2007). The role of GT-richness in germ-line recombination is
less clear (Majewski and Ott, 2000; Wahls, 1998).
Genetic studies that relate high linkage to low recombination
between polymorphic loci indicate that in recombination hot spots
"there exist multiple fuzzy DNA sequence determinants … based on the
nature of the allele present" (Nishant and Rao, 2006).
Our approach is less specific in that we study DNA, as DNA,
irrespective of the underlying function. |
|
For a given duplex DNA segment, the potential for a strand to be extruded to adopt a stem-loop structure can be quantitated as the "folding of natural sequence" (FONS) value of that structure. This, in turn, can be decomposed into a base order-dependent component (quantitated as the "folding of randomized sequence difference;" FORS-D) and a base composition-dependent component (quantitated as the "folding of randomized sequence mean;" FORS-M). These terms refer to methods of calculation from the energy values of computer-folded sequences (Mathews and Turner, 2006; Zuker, 2000). The average value of several independently shuffled versions of a sequence (base order having thus been eliminated) provides the base composition-dependent component. This value is subtracted from the corresponding value for the total folding energy (FONS) to obtain the base order-dependent component. Whatever the relative contributions of base order and composition, when the FONS values of top and bottom strands are close to zero, classical duplex forms would be expected to predominate in a DNA segment. High negative values would be expected to stabilize extruded stem-loops, a process that would be favoured by negative supercoiling (Forsdyke, 1995b; 1998; Wang et al., 1990). Misconceptions concerning the shuffling of sequences at the dinucleotide level, rather than at the level of single bases (mononucleotides), are discussed elsewhere (Forsdyke, 2007b; Xu et al., 2007). |
5. Symmetrical distribution of folding energies of top and bottom strands
|
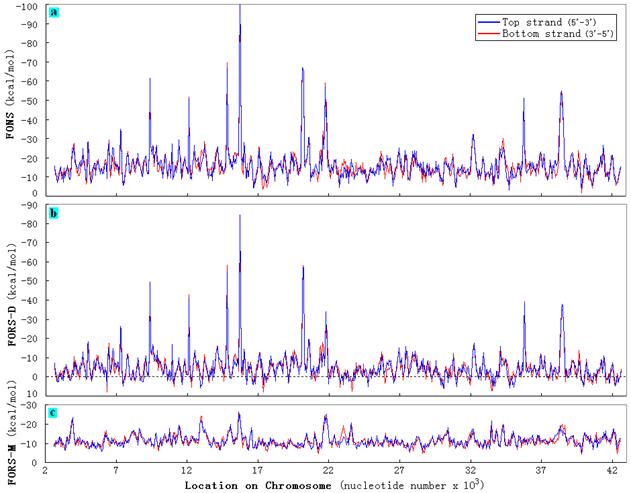
Folding energy values were determined for 200 nt segments ("windows") from the genome of C. elegans that were moved in steps of 50 nt along a sequence, using our program "Random_fold_scan," with base order being randomized at the mononucleotide level by shuffling (Xu et al., 2007; Zhang et al., 2008). A representative result is shown in Figure 1. |
|
|
| Figure 1. The distribution of values for (a) FONS (total energy of folding), (b) FORS-D (the base order-dependent component), and (c) FORS-M (the base composition-dependent component), in the top strand (blue line) and bottom strand (red line) of a 40 kb segment of chromosome I of C. elegans (nucleotides 2500 to 42500). Folding energies were calculated for sequence windows (200 nt) that were moved in 50 nt steps. Sequences were retrieved from GenBank (accession numbers: chromosome I: NC_003279.5; chromosome II: NC_003280.6; chromosome III: NC_003281.7; chromosome IV: NC_003282.4; chromosome V: NC_003283.7; chromosome X: NC_003284.6). |
|
Total folding energy values fluctuated widely (Fig. 1a), and these fluctuations reflected changes in the base order-dependent component of the folding energy (Fig. 1b) more than the base composition-dependent component (Fig. 1c). The folding energies of top and bottom strands followed each other closely. However, there were small differences. Sometimes the top strand had the greatest folding potential (more negative FONS value), and sometimes the bottom strand had the greatest folding potential. The similarity between the two strands was also reflected in the values for FORS-D (Fig. 1b) and FORS-M (Fig. 1c). As described previously for various biological species, whereas FONS and FORS-M values were always negative, some FORS-D values were positive. Sometimes base order and base composition work together to generate the total folding energy (FONS) value and sometimes they work in opposition (Forsdyke, 1998; Xue and Forsdyke, 2003) .
To investigate other chromosomes,
600 windows (200 nt) were randomly selected from each chromosome
(software for this selection is available on request). Table
1 shows that no significant overall differences were
observed between the mean FORS-D values of top and bottom strands of
each chromosome. Whereas the autosomes (chromosomes I to V)
had mean values around -4.32 kcal/mol,
the X chromosome had a significantly lower
mean value (-2.35 kcal/mol; P<0.0001). Thus, on average, the X
chromosome of C. elegans displayed
lower base order-dependent stem-loop potential than autosomes. For
each chromosome the average of the absolute differences between
individual paired FORS-D values of top and bottom strands was
significantly different from zero (Table
1; P<0.0001).
The average X chromosome absolute FORS-D difference
(1.74 kcal/mol) resembled that of chromosome I (1.73
kcal/mol), but differed significantly from the other autosomes
(average difference 4.23 kcal/mol; P<0.0001). |
|
Folding
energies (kcal/mol) |
Chromosome |
||||||
|
1 |
2 |
3 |
4 |
5 |
X |
||
|
FORS-D |
Top strands (mean) |
-4.73 |
-3.97 |
-5.14 |
-4.23 |
-3.86 |
-2.29 |
|
Bottom strands
(mean) |
-4.70 |
-3.94 |
-4.88 |
-4.10 |
-3.68 |
-2.42 |
|
|
Probability (P) a |
0.71 |
0.90 |
0.24 |
0.55 |
0.39 |
0.17 |
|
|
Absolute
differences (mean) b |
1.73±0.06 |
4.14±0.14 |
4.21±0.13 |
4.47±0.13 |
4.10±0.12 |
1.74±0.06 |
|
|
FORS-M |
Top strands (mean) |
-11.34 |
-22.73 |
-22.91 |
-22.43 |
-22.65 |
-11.12 |
|
Bottom strands (mean) |
-11.43 |
-22.98 |
-22.67 |
-22.41 |
-22.54 |
-11.13 |
|
|
Probability (P) a |
0.18 |
0.19 |
0.19 |
0.92 |
0.56 |
0.85 |
|
|
Absolute differences (mean) b |
1.21±0.04 |
3.77±0.11 |
3.68±0.11 |
3.51±0.11 |
3.72±0.11 |
1.04±0.03 |
|
a The
significance of differences between top and bottom strand means (paired t-test).
b Absolute
differences between individual values for top and bottom strands. Values
are presented together with the standard error of the mean.
6.
Asymmetrical distribution of folding energies in telomeres
|
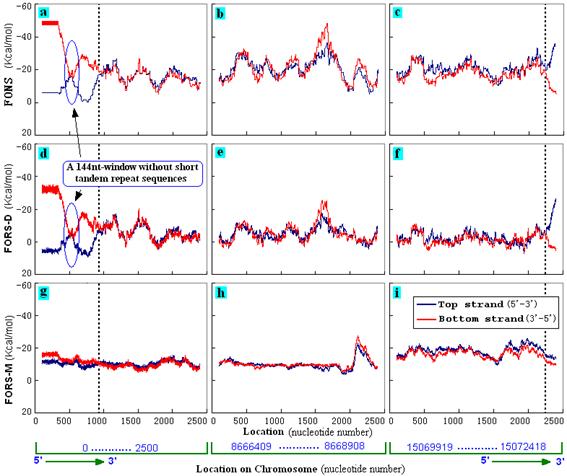
The symmetry of folding energies between the two strands was lost in telomeric regions which contain STRs (Cangiano and La Volpe, 1993). Figure 2 compares the two telomeric regions of chromosome I with a region randomly selected from the middle of the chromosome. In the telomeric regions the strands containing GT-rich repeats (bottom strand in Fig. 2a and top strand in Fig. 2c) had greater total folding energy (FONS) than the AC-rich complementary strands. This was due to differences in base order (Figs. 2d, f) more than in base composition (Figs. 2g, i). In contrast, relative symmetry was displayed by a short 144 nt segment in a telomeric region that was devoid of GT-rich STRs (Figs. 2a, d, g), and by the central non-telomeric region (Figs. 2b, e, h). Similar results were obtained with the telomeres of the other five C. elegans chromosomes (data not shown). |
|
|
|
Figure 2. Telomeres
display strand asymmetry. Segments
(2.5 kb) from the 5' end (a, d, g), middle (b, e, h) and 3' end (c,
f, i) of chromosome I of C. elegans were
folded as in Figure 1 to determine FONS values (a, b, c), FORS-D
values (d, e, f), and FORS-M values (g, h, i). Telomeric boundaries
are indicated by the vertical dashed line. For further details
please see text. |
|
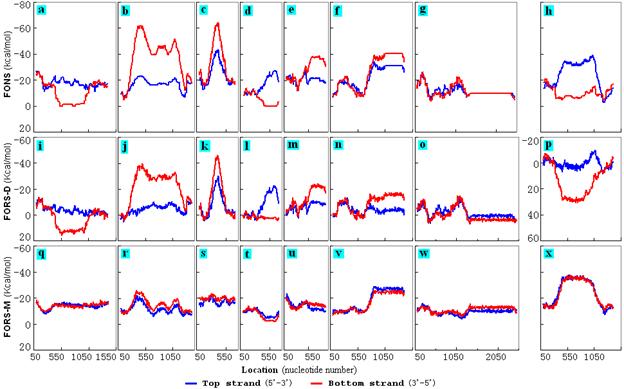
The symmetry of folding energies between the two strands was also lost in many internal regions containing STRs (with repeat unit sizes starting at 2 nt). A sample of these regions was obtained from chromosomes I and II using Tandem Repeats Finder (Benson, 1999). STR-containing segments were required to have individual repeats that (i) were short (less than 13nt), (ii) closely matched the other repeats in the segment (>90%), and (iii) had a collective length of at least 200nt. These were selected for fold analysis together with their flanking sequences. The results showed that some, but not all, of these internal STR sequences displayed the asymmetry in FONS (Figs. 3 a-f, h). A region on chromosome II (bases 6183938-6186538), which contained the 7nt-STR unit ACGCTAT that had a 100% match with its companion repeats and only slightly violated Chargaff's second parity rule, did not display the asymmetry (Fig. 3g). Sometimes the top strand displayed the greatest folding propensity (more negative FONS value; Figs. 3 a, d, h), and sometimes the bottom strand (Figs. 3 b, c, e, f). Again, the asymmetries reflected differences in base order (FORS-D; Figs. 3i-p) more than in base composition (FORS-M; Figs. 3q-x). |
|
|
|
Figure 3. Internal
STR-containing sequences display strand asymmetry. Fold analysis of
some segments of C. elegans chromosomes
I and II that contain internal STR sequences. These segments are
from chromosome I: 748813-750313 (a, i, q), 2254662-2256162 (b, j,
r), 2254662-2256162 (c, k, s), 11655315-11656815 (h, p, x), and
chromosome II: 2000479-2001279 (d, l, t), 2631644-2632644 (e, m, u),
3270800-3272300 (f, n, v), 6183938-6186538 (g, o, w), respectively.
They were folded as
in Figure 1 to determine FONS values (a-h), FORS-D values (i-p) and
FORS-M values (q-x). |
8.
Asymmetrical distribution is greater with short repeat units
|
Rather than continue with natural
sequences, to identify general sequence characteristics that might
confer large differences in folding values between top and bottom
strands, we generated an artificial set of top strand STR-containing
sequences that varied in repeat unit length (5 - 50
nt), base composition, and base order. To avoid
duplication in or between different groups, these
STR sequences had to meet the following criteria: (i) Ability to
fold using RNAstructure 4.2 with local data files for DNA (Mathews
and Turner, 2006). (ii) Absence of internal tandem repeats. A repeat
unit such as 5'ATGAATGA3' would not qualify since it contains two
shorter repeat units. (iii) No cyclic permutability. For all
possible tandem repeat sequences with the same base order but
different start nucleotides (e.g. 5' GGACTAAT3', 5' GACTAATG3', 5'
ACTAATGG3' and 5' CTAATGGA3'), only one can be selected. (iv) No
reverse complementarity. If two sequences are reverse complementary
(e.g. 5' GGACTAAT 3' and 5'ATTAGTCC 3'), only one can be selected. A
program (written with ActivePerl-5.8.8.820)
that can generate these sequences when given the size of repeat
units and their duplication number, is available on request.
Previous studies having shown the utility of 200nt
windows sizes, here we used the program to generate
STRs of this size. A
200 nt sequence window might contain 20 copies of a 10 nt repeat
unit, or 4 copies of a 50 nt repeat unit. For a
given size of repeat unit a total of 600 STRs were generated
randomly. For 5nt and 6nt repeat units only 58 and 220 sequences,
respectively, satisfied the above criteria. STRs with smaller repeat
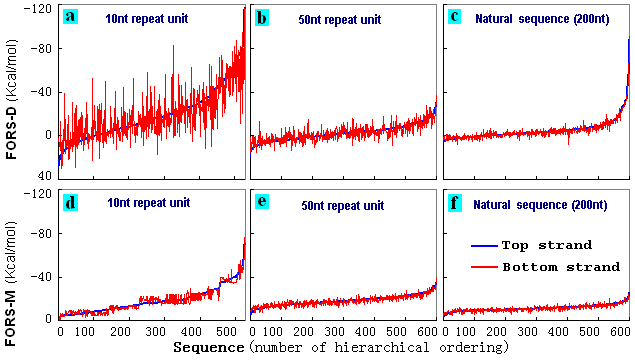
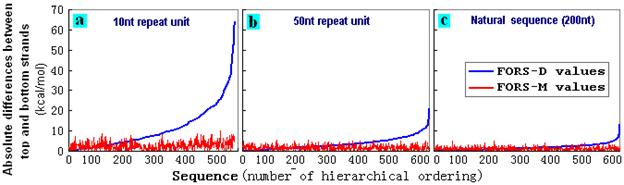
units were not generated. 600 top strand FORS-D values for such computer-generated STRs were hierarchically ordered and plotted with the corresponding bottom strand values (Fig. 4). In some cases the bottom strand values were more negative than top strand values. In other cases the bottom strand values were less negative than top strand values. Thus, while the top strand values gave smooth lines (because of the hierarchical ordering), bottom strand values gave irregular lines. These irregular fluctuations were greater in the case of STRs with shorter subunits (Fig. 4a), and were least in the controls (a set of natural sequences; Fig. 4c). Studies with other subunit sizes indicated progressively greater fluctuations as subunit size decreased (data not shown). The asymmetries were much less evident in the case of FORS-M values (Figs. 4d-f). |
|
|
|
Figure 4. Variation
of bottom strand folding values (irregular red line) relative to top
strand folding values (regular blue line) for a series of 600
computer-generated STRs. Top strand
values for FORS-D (a, b, c) and FORS-M
(d, e, f) were hierarchically ordered from low negative to high
negative, and plotted with the corresponding bottom strand values.
The 200 nt STR sequences in (a) and (d) have 20 x 10-nt repeats;
those on (b) and (e) have 4 x 50-nt repeats. The 600 natural 200
nt sequences, (c) and (f), were randomly selected from chromosome I
of C. elegans. |
|
The signs of the fluctuations were eliminated by taking absolute differences between top and bottom strand FORS-D values, which were again hierarchically ordered (Fig. 5). When compared with corresponding absolute FORS-M differences, that were relatively constant, absolute FORS-D differences of STR sequences exhibit larger values and great variation (Figs. 5a, b). |
|
|
| Figure 5. Variation of absolute FORS-M differences (irregular red line) relative to absolute FORS-D differences (regular blue line) for a series of computer-generated STRs. The values of absolute FORS-D differences were hierarchically ordered from low to high, and plotted with the corresponding values of absolute FORS-M differences. The STR sequences in (a) and (b) are 20 x 10-nt repeats and 4 x 50-nt repeats, respectively. The natural 200 nt sequences (c) are randomly selected from chromosome I of C. elegans. |
9.
Asymmetrical distribution when second parity rule is violated
|
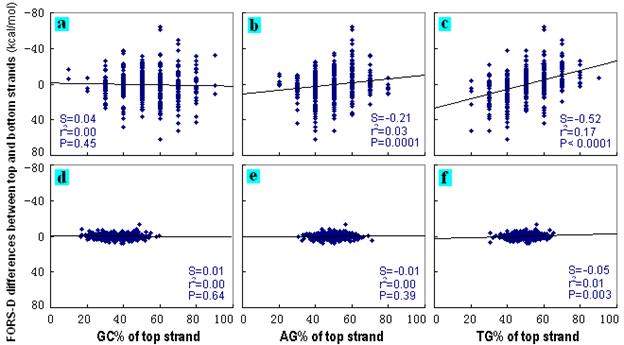
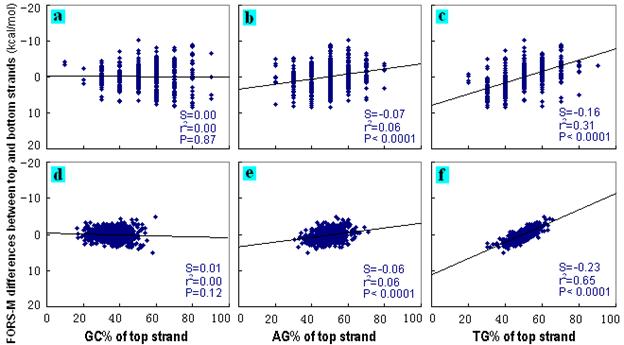
For 600 computer-generated 10 nt
repeat unit STRs, the influence of top strand base composition (e.g.
[G + C]% written as GC%) on the difference between top and bottom
strand FORS-D and FORS-M values was investigated by first order
linear regression. Differences in GC% (i.e. Chargaff's second parity
rule was not violated) affected the differences between the strands
neither of the STR-containing sequences (Figs. 6a, 7a), nor of the
control series of 600 natural 200 nt sequences (Figs. 6d, 7d). Thus,
there were no differences in FONS values (the sum of FORS-D and
FORS-M values). Stem-loop extrusion from duplexes would be
symmetrical both at low and high GC% values.
However, differences in AG% (that
would reciprocally correspond to bottom strand CT%), and especially
differences in GT% (that would reciprocally correspond to bottom
strand AC%), were correlated with large differences between both the
FORS-D values and FORS-M values in the case of the
computer-generated STR-containing sequences (Figs. 6b,c; Figs.
7b,c). Thus, this group of sequences included sequences that, by
virtue both of their base composition and order, would have differed
widely in the FONS values of their complementary strands. Stem-loop
extrusion from such duplexes would have been increasingly
asymmetrical as their A+G or G+T content increased.
In the case of the natural
sequences, differences in AG% and GT% either did not correlate with
differences in strand FORS-D values (Fig. 6e), or correlated very
weakly (Fig. 6f). On the other hand, differences in AG%, and
especially differences in GT%, correlated well with differences in
strand FORS-M values (Figs. 7e, f). Thus, total folding energy
values (FONS) would differ between top and bottom strands as the A+G
or G+T content of the top strand of a segment increased, but this
would mainly reflect differences in base composition.
The particular computer-generated sequences which, by virtue of
their base orders, accounted for the differences
between FORS-D values (Figs. 6b,c) would either not have been
present, or would have been present at very low frequencies, in the
natural sequences (Figs. 6e,f). Thus, for natural sequences it is
usually differences in base composition alone (AG% or GT%), that
correlate with asymmetrical folding of top and bottom strands.
Differences in GC% do not violate the second parity rule so folding
is symmetrical. |
|
|
|
Figure 6. Dependence
of FORS-D differences between top and bottom strands on base
compositions. The 600 FORS-D differences for computer-generated
200nt sequences containing tandem 10nt repeat units were plotted
against the corresponding values for (a) GC%, (b) AG% and (c) GT%.
The 600 FORS-D differences for randomly selected natural 200nt
sequences from chromosome I were plotted similarly (d, e, f). Base
compositions refer to the top strand of each DNA sequence. FORS-D
differences were calculated by substracting bottom strand values
from the corresponding top strand values. Parameters of the
least-squares regression lines are slope (S), the coefficient of
determination (r2),
and the probability (P value) that the slope of the line is not
significantly different from zero. Statistical
analyses were conducted with GraphPad Prism version 4.03 for
Microsoft Windows. |
|
|
|
Figure 7.
Dependence of FORS-M differences between top and bottom strands on
base compositions. For details see Figure 6. |
|
For most sequence segments violations of Chargaff's second parity rule are minimal, and the strands of a duplex have the potential to adopt single-strand stem-loop configurations in a symmetrical manner (Fig. 1; Table 1). In this circumstance, recombination could be in accord with models for germ-line meiotic strand pairing based on Crick's unpairing postulate. The major disruptor of the pairing between two independent duplexes would be differences in GC% between the two duplexes, the individual strands of each duplex being extruded symmetrically by virtue of their common GC% (Forsdyke, 2006; 2007a). Where symmetry failed it would be due to strand differences in certain bases (i.e. AG% and/or GT%), irrespective of their order (Figs. 6, 7). Thus, distinct sequences would not be involved. However, the symmetry would fail in telomeric regions (Fig. 2) and in other regions containing STRs (Fig. 3), due to both the composition and the order of bases (Figs. 6, 7). Distinct sequences would be involved. The failure would be greatest when the STRs were small and violations of Chargaff's second parity rule were maximal (Figs. 4, 5; Table 2). |
Table 2.
Linear regression analysis of FORS-D differences between top and bottom strands
versus base composition of STRs
|
Base composition a |
Repeat units
contained in 200 nt STRs b |
|||||||||
|
5nt |
6nt |
7nt |
10 nt |
13 nt |
20 nt |
30 nt |
40 nt |
50 nt |
||
|
GC% |
S |
0.0726 |
-0.0013 |
0.0018 |
0.0354 |
-0.0233 |
0.0703 |
-0.0019 |
0.0072 |
0.0483 |
|
r2 |
0.0031 |
0.0000 |
0.0000 |
0.0011 |
0.0005 |
0.0063 |
0.0000 |
0.0001 |
0.0036 |
|
|
P |
0.6970 |
0.9875 |
0.9677 |
0.4494 |
0.5860 |
0.0572 |
0.9561 |
0.8135 |
0.1429 |
|
|
AG% |
S |
-0.5945 |
-0.5485 |
-0.2338 |
-0.2079 |
-0.2065 |
-0.0885 |
-0.0832 |
-0.0566 |
0.0076 |
|
r2 |
0.0607 |
0.1097 |
0.0367 |
0.0281 |
0.0388 |
0.0095 |
0.0096 |
0.0060 |
0.0001 |
|
|
P |
0.0813 |
< 0.0001 |
< 0.0001 |
0.0001 |
< 0.0001 |
0.0193 |
0.0175 |
0.0584 |
0.8094 |
|
|
GT% |
S |
-1.0890 |
-0.5868 |
-0.4742 |
-0.5215 |
-0.4139 |
-0.1989 |
-0.2039 |
-0.0884 |
-0.0945 |
|
r2 |
0.4250 |
0.2117 |
0.1807 |
0.1748 |
0.1393 |
0.0477 |
0.0587 |
0.0134 |
0.0151 |
|
|
P |
< 0.0001 |
< 0.0001 |
< 0.0001 |
< 0.0001 |
< 0.0001 |
< 0.0001 |
< 0.0001 |
0.0047 |
0.0026 |
|
a S,
slope; r2, the coefficient of determination; P,
the probability that the slope of the least-squares line is not significantly
different from zero.
b For
5nt and 6nt repeat unit sizes, only 58 and 220 sequences could be generated by
our program.
|
In view of the highly specialized nature of telomeres (Verdun and Karlseder, 2007) and the fact that asymmetry between top and bottom strands can be associated with specialized forms of recombination (Huang et al., 2007), it seems likely that telomeric and interspersed regions containing STRs (Figs. 2, 3) are, in some way, specialized for functions involving somatic recombination, and contain distinct sequences adapted for this. In such regions recombination would be highly regulated, requiring specialized proteins analogous to some of those mediating recombination in the GT-rich islands containing Chi sequences in E. coli.
For example, the
telomeric DNA of eukaryotes such as C. elegans has
a single-stranded GT-rich 3' extension that would only weakly bond
with itself. As such, under the influence of a variety of
specialized proteins (Raices et al., 2008) it
might readily invade a neighboring duplex with its identical strand
being displaced as a "D-loop." The resulting "T-loop" might decrease
telomere erosion. Alternatively, the single stranded form might more
readily engage in a recombination-dependent form of telomere
regeneration known as ALT ("alternative lengthening of telomeres"; Verdun
and Karlseder, 2007). It should be noted that, depending on salt
concentration, G-rich regions have the potential (i) to aggregate (Forsdyke,
1984) with the formation of G-quartets (Henderson
et al., 1987; Williamson et al., 1989) , and (ii) to
form Z-DNA (Haniford and Pulleyblank, 1983). The
folding programs we have employed (Mathews and Turner,
2006; Zuker, 2000) do not take such possibilities into
account. Apart from a somatic role, their association with meiotic recombination hotspots suggests a role for STRs in the germ line. That the association is indicative of a role in the termination, rather than initiation, of meiotic recombination has been suggested by Wahls (1998); some characteristic of satellites is held to arrest the branch migration that may follow formation of Holliday junctions. Thus, recombination could initiate in a flanking non-satellite region and terminate within the satellite due to some inhibitory influence. Perhaps the enzymes involved would sense the potential extrusion asymmetry.
In summary,
extrusions of higher ordered structures from DNA duplexes vary
between the extremes of close symmetry and highly asymmetry. We have
argued that a germ-line event (the initiation of divergence into
distinct species) is likely to be influenced by meiotic
recombination when there is symmetry of strand extrusion from DNA
duplexes (i.e. Chargaff's second parity rule applies). If this is
true then, by virtue of their extrusion asymmetry, regions adapted
for special forms of somatic recombination might be less favorably
adapted for the strand pairing that initiates meiotic recombination
(for an alternative view see Rockmill and Roeder, 1998). High
variability (associated with microsatellite content) might then fail
to register in terms of the increased pairing incompatability
between homologous chromosomes that is expected to precede
speciation. In this circumstance, and without excluding roles for
other classes of repeat sequence, a role for microsatellites as
drivers of speciation is in doubt. |
Acknowledgements
This work was supported by
research grants from National Natural Science Foundation of China (No. 30600352)
and Natural Science Foundation of Jiangsu Province, China (No. BK2006550), and
the Startup Fund from
Allawi, H.T., and SantaLucia, J., Jr., 1998.
NMR solution structure of a DNA dodecamer containing single G.T mismatches.
Nucleic Acids Res. 26, 4925-4934.
Baldwin, G.S., Brooks, N.J.,
Robson, R.E., Wynveen, A., Goldar, A., Leikin, S., Seddon, J.M., and Kornyshev,
A.A., 2008. DNA Double Helices Recognize Mutual Sequence Homology in a Protein
Free Environment. J. Phys. Chem. B 112, 1060-1064.
Bell, S.J., Chow, Y.C., Ho,
J.Y., and Forsdyke, D.R., 1998. Correlation of chi orientation with
transcription indicates a fundamental relationship between recombination and
transcription. Gene 216, 285-292.
Benson, G., 1999. Tandem
repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27,
573-580.
Britten, R.J., and Davidson,
E.H., 1971. Repetitive and non-repetitive DNA sequences and a speculation on the
origins of evolutionary novelty. Quart. Rev. Biol. 46, 111-138.
Cangiano, G., and La Volpe, A.,
1993. Repetitive DNA sequences located in the terminal portion of the Caenorhabditis
elegans chromosomes. Nucleic Acids Res. 21, 1133-1139.
Crick, F., 1971. General model
for the chromosomes of higher organisms. Nature 234, 25-27.
Crick, F., 1974. Letter from F.
Crick to G. Khorana 28th June 1974. National Library of Medicine, Washington. http://profiles.nlm.nih.gov/SC/B/B/M/Q/_/scbbmq.pdf.
Crowther, C.R., 1922.
Evolutionary faith and modern doubts. Nature 109, 777.
Doyle, G.G., 1978. A general
theory of chromosome pairing based on the palindromic DNA model of Sobell with
modifications and amplifications. J. Theor. Biol. 70, 171-184.
Ellegren, H., 2004.
Microsatellites: simple sequences with complex evolution. Nat. Rev. Genet. 5,
435-445.
Flamm, W.G., Walker, P.M., and
McCallum, M., 1969. Some properties of the single strands isolated from the DNA
of the nuclear satellite of the mouse (Mus musculus). J. Mol. Biol. 40, 423-443.
Flavell, R.B., Sequence
amplification, deletion and rearrangement: major sources of variation during
species divergence, in: Dover, G. A., Flavell, R.B. , (Ed.), Genome Evolution, Academic
Press, San Diego 1982, pp. 301-323.
Forsdyke, D.R., 1984.
Purification of oligo dG-tailed Okayama-Berg linker DNA fragments by oligo
dC-cellulose chromatography. Anal. Biochem. 137, 143-145.
Forsdyke, D.R., 1995a. Relative
roles of primary sequence and (G + C)% in determining the hierarchy of
frequencies of complementary trinucleotide pairs in DNAs of different species.
J. Mol. Evol. 41, 573-581.
Forsdyke, D.R., 1995b.
Reciprocal relationship between stem-loop potential and substitution density in
retroviral quasispecies under positive Darwinian selection. J. Mol. Evol. 41,
1022-1037.
Forsdyke, D.R., 1998. An
alternative way of thinking about stem-loops in DNA. A case study of the human
G0S2 gene. J. Theor. Biol. 192, 489-504.
Forsdyke, D.R., 2006.
Evolutionary Bioinformatics. Springer, New York.
Forsdyke, D.R., 2007a.
Molecular sex: the importance of base composition rather than homology when
nucleic acids hybridize. J. Theor. Biol. 249, 325-330.
Forsdyke, D.R., 2007b.
Calculation of folding energies of single-stranded nucleic acid sequences:
Conceptual issues. J. Theor. Biol. 248, 745-753.
Forsdyke, D.R., and Mortimer,
J.R., 2000. Chargaff's legacy. Gene 261, 127-137.
Gierer, A., 1966. Model for DNA
and protein interactions and the function of the operator. Nature 212,
1480-1481.
Haniford, D.B., and
Pulleyblank, D.E., 1983. Facile transition of poly[d(TG) x d(CA)] into a
left-handed helix in physiological conditions. Nature 302, 632-634.
Henderson, E., Hardin, C.C.,
Walk, S.K., Tinoco, I., Jr., and Blackburn, E.H., 1987. Telomeric DNA
oligonucleotides form novel intramolecular structures containing guanine-guanine
base pairs. Cell 51, 899-908.
Huang, F.T., Yu, K., Balter,
B.B., Selsing, E., Oruc, Z., Khamlichi, A.A., Hsieh, C.L., and Lieber, M.R.,
2007. Sequence dependence of chromosomal R-loops at the immunoglobulin
heavy-chain Smu class switch region. Mol. Cell. Biol. 27, 5921-5932.
Kleckner, N., and Weiner, B.M.,
1993. Potential advantages of unstable interactions for pairing of chromosomes
in meiotic, somatic, and premeiotic cells. Cold Spring Harb. Symp. Quant. Biol.
58, 553-565.
Lao, P.J., and Forsdyke, D.R.,
2000. Crossover hot-spot instigator (Chi) sequences in Escherichia coli occupy
distinct recombination/transcription islands. Gene 243, 47-57.
Lauffer, M.A., 1975.
Entropy-Driven Processes in Biology. Springer-Verlag, New York.
Leach, D.R., 1994. Long DNA
palindromes, cruciform structures, genetic instability and secondary structure
repair. Bioessays 16, 893-900.
Majewski, J., and Ott, J.,
2000. GT repeats are associated with recombination on human chromosome 22.
Genome Res. 10, 1108-1114.
Mathews, D.H., and Turner,
D.H., 2006. Prediction of RNA secondary structure by free energy minimization.
Curr. Opin. Struct. Biol. 16, 270-278.
Muller, H.J., 1941. Resumé and
perspectives of the symposium on genes and chromosomes. Cold Spring Harb. Symp.
Quant. Biol. 9, 290-308.
Nishant, K.T., and Rao, M.R.,
2006. Molecular features of meiotic recombination hot spots. Bioessays 28,
45-56.
Orgel, L.E., and Crick, F.H.,
1980. Selfish DNA: the ultimate parasite. Nature 284, 604-607.
Phillips, G.J., Arnold, J., and
Ivarie, R., 1987. Mono- through hexanucleotide composition of the Escherichia
coli genome: a Markov chain analysis. Nucleic Acids Res. 15,
2611-2626.
Prabhu, V.V., 1993. Symmetry
observations in long nucleotide sequences. Nucleic Acids Res. 21, 2797-2800.
Raices, M., Verdun, R.E.,
Compton, S.A., Haggblom, C.I., Griffith, J.D., Dillin, A., and Karlseder, J.,
2008. C. elegans telomeres contain G-strand and C-strand overhangs that are
bound by distinct proteins. Cell 132, 745-757.
Robertson, M., 1981. Gene
families, hopeful monsters and the selfish genetics of dna. Nature 293, 333-334.
Rockmill, B., and Roeder, G.S.,
1998. Telomere-mediated chromosome pairing during meiosis in budding yeast.
Genes Dev. 12, 2574-2586.
Rogerson, A.C., 1989. The
sequence asymmetry of the Escherichia coli chromosome
appears to be independent of strand or function and may be evolutionarily
conserved. Nucleic Acids Res. 17, 5547-5563.
Sobell, H.M., 1972. Molecular
mechanism for genetic recombination. Proc. Natl. Acad. Sci. USA 69, 2483-2487.
Tracy, R.B., Chedin, F., and
Kowalczykowski, S.C., 1997. The recombination hot spot chi is embedded within
islands of preferred DNA pairing sequences in the E. coli genome. Cell 90,
205-206.
Verdun, R.E., and Karlseder,
J., 2007. Replication and protection of telomeres. Nature 447, 924-931.
Wagner, R.E., Jr., and Radman,
M., 1975. A mechanism for initiation of genetic recombination. Proc. Natl. Acad.
Sci. USA 72, 3619-3622.
Wahls, W.P., 1998. Meiotic
recombination hotspots: shaping the genome and insights into hypervariable
minisatellite DNA change. Curr. Top. Dev. Biol. 37, 37-75.
Wang, J.C., Caron, P.R., and
Kim, R.A., 1990. The role of DNA topoisomerases in recombination and genome
stability: a double-edged sword? Cell 62, 403-406.
Watson, J.D., and Crick, F.H.,
1953. Genetical implications of the structure of deoxyribonucleic acid. Nature
171, 964-967.
Williamson, J.R., Raghuraman,
M.K., and Cech, T.R., 1989. Monovalent cation-induced structure of telomeric
DNA: the G-quartet model. Cell 59, 871-880.
Xu, S.G., Wei, J.F., and Zhang,
C.Y., 2007. A FORS-D analysis software "Random_fold_scan" and the influence of
different shuffle approaches on FORS-D analysis [in Chinese]. J. Jiangsu
Univ.(Med. Edition) 17, 461-466,470.
Xue, H.Y., and Forsdyke, D.R.,
2003. Low-complexity segments in Plasmodium falciparum proteins are primarily
nucleic acid level adaptations. Mol. Biochem. Parasitol. 128, 21-32.
Zhang, C.Y., Wei, J.F., and He,
S.H., 2005a. The key role for local base order in the generation of multiple
forms of China HIV-1 B'/C intersubtype recombinants. BMC Evol. Biol. 5, 53.
Zhang, C.Y., Wei, J.F., and He,
S.H., 2005b. Local base order influences the origin of ccr5 deletions mediated
by DNA slip replication. Biochem. Genet. 43, 229-237.
Zhang, C.Y., Wei, J.F., Wu,
J.S., Xu, W.R., Sun, X., and He, S.H., 2008. Evaluation of FORS-D Analysis: A
Comparison with the Statistically Significant Stem-loop Potential. Biochem.
Genet. 46, 29-40.
Zickler, D., 2006. From early
homologue recognition to synaptonemal complex formation. Chromosoma 115,
158-174.
Zuker, M., 2000. Calculating
nucleic acid secondary structure. Curr. Opin. Struct. Biol. 10, 303-310.
![]()
Bioinformatics Index (Click Here)
HomePage (Click Here)
![]()
This page was established in May 2008 and was last edited on 14 Aug 2008 by D. Forsdyke