|
|
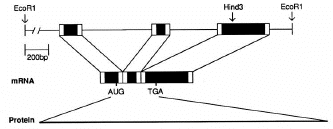
| Splicing of the human G0S19 (MIP1-alpha/CCL3) gene to form messenger RNA, part of which is then translated to form the chemokine protein whose receptor (CCR5) is also a HIV1 coreceptor. |
Are Introns In-series Error-detecting Sequences? (Forsdyke 1981)
Introns, Antirecombination and GC% (Matsuo et al. 1994)
Alternative Introns Early Theory (Forsdyke 1994)
A Stem-Loop "Kissing" Model (Forsdyke 1994, 1995a)
Conservation of Stem-Loop Potential in Introns (Forsdyke 1995b)
Stem-Loop Potential in Retroviruses (Forsdyke 1995c)
Stem-loop potential in MHC genes (Positive Darwinian Selection; Forsdyke 1996)
Stem-loop potential in heat-shock genes (Forsdyke 1995-8)
Evidence from Overlapping Genes (Barrette et al. 2001)
Positive Darwinian Selection using a Single Sequence (Forsdyke 2007)
Videos on Exons and Introns 2012
|
In the 1960s non-bacterial (eukaryotic) ribosomal RNAs (rRNAs) were found to be synthesized as a long precursor RNA which was subsequently processed by the removal of apparently functionless internal "spacer" sequences. Since bacterial (prokaryotic) rRNAs were more compactly organized, it was reasonable to ask whether the first rRNAs to evolve had the spacer sequences, which subsequently decreased in prokaryotes, or whether the spacer sequences were later acquired in eukaryotes. In the 1960s a similar processing was found to apply to eukaryotic precursor messenger RNAs (pre-mRNAs; Scherrer et al. 1970). In the mid 1970s it was found that the some of the internal sequences interrupted the protein-encoding part of the corresponding mRNAs. The internal sequences which were removed were named "introns", and what remained in the processed mRNA constituted the "exons". Since the phenomenon had already been described for rRNA it should have been no big deal to find that it also applied to other RNAs, but many, including the author of these pages, were surprised that protein-encoding regions were interrupted. The same questions remained.
If introns could be dispensed with in bacteria, then perhaps they had no function. Alternatively, whatever function introns had, either was not necessary in bacteria, or might be achieved in other ways by bacteria. Since members of many bacterial species appeared to be under intense pressure to streamline their genomes to facilitate rapid replication, if it were possible they would have dispensed with any preexisting introns and/or would have been reluctant to acquire them. On the other hand, if introns played a role and/or did not present too great a selective burden, eukaryotes would have tended to retain preexisting introns, or could have acquired them (Raible et al. 2005). Knowing the function of introns seemed critical for sorting out these issues. There were many ingenious suggestions. Some thought introns were just another example of the apparently non-utile "junk" DNA which littered the DNA of many eukaryotes. However, some principles to guide investigation of a possible error-checking role were presented (Forsdyke 1981; Reanney 1984). There is now growing evidence that introns play such a role (Forsdyke 1995a,b; Barrette et al. 2001), although the mechanism may be somewhat different to that originally proposed (Liebovitch et al. 1996. Biophys. J. 71, 1539-1544). It appears that the order of bases in nucleic acids might have been under evolutionary pressure to develop the potential to form stem-loop structures which would facilitate "in-series" or "in-parallel" error-correction by recombination. Although the genetic code is degenerate (more than one codon per amino acid so that there is some flexibility as to which base occupies a particular position), there is still room for conflict between the "desires" of a sequence to encode both a protein (or non-messenger RNA) and stem-loop potential. The conflict would be particularly apparent in the case of genes under very strong positive phenotypic (Darwinian) selection, as in the case of genes affected by "arms races" with predators or prey. For example, snake venom may decrease the rodent population (prey) until a venom-resistant rodent line develops and expands. Now, while the rodent population increases, the snake population (predators) decreases, because it cannot obtain sufficient food. This decrease continues until a line of snakes arises with more active venom, which overcomes the resistance. This line of snakes now increases, and the rodent population begins to fall again. This cycle constitutes an "arms race", and influences particular genes. The part of the venom protein which is important for toxicity is required to change so very rapidly in response to this strong phenotypic pressure from the environment, that the corresponding gene can no longer afford the "luxury" of trying to encode both the best protein and the best stem-loops. So the stem-loop role is left to the introns. Here, paradoxically, sequence conservation is high, whereas in the exons, sequence conservation is low (Forsdyke 1995b). Similar pressures may be acting on the peptide binding regions of the genes encoding major histocompatibility antigens (Forsdyke 1996b), and affect overlapping genes (Barrette et al. 2001). There is another "player" in the conflict between protein-encoding potential and stem-loop potential. Most mRNAs are "purine-loaded" in the loop regions of stem-loop structures (reviewed in Forsdyke & Mortimer, 2000). The selection pressure for this appears to operate primarily at the cytoplasmic level. Consequently, the purine-loading may not optimally serve the postulated genomic role of stem-loops. There may be a conflict between "AG-pressure" (the pressure to purine-load) and stem-loop pressure. To resolve this, stem-loop potential would be moved to a region where AG-pressure does not operate, the introns. Scherrer, K. et al. (1970) Nuclear and cytoplasmic messenger-like RNA and their relation to the active messenger RNA in polyribosomes of HeLa cells. Cold Spring Harb. Symp. Quant. Biol. 35, 539-554. Raible, F. et al. (2005) Vertebrate-type intron-rich genes in the murine annelid Platynereis dumerilii.Science 310, 1325-6. Reanney D. C. (1984) RNA splicing as an error-screening mechanism. Journal of Theoretical Biology 111, 315-321. Donald Forsdyke circa 2000 |
![]()
Are Introns In-series Error-detecting Sequences?
DONALD R. FORSDYKE
The Journal of Theoretical Biology (1981) 93, 861-866 [With the permission of the copyright holder, Academic Press]
(Received 17 December 1980, and in revised form 28 May 1981)
| Summary. The probability of the accurate transmission of a message sequence can be increased by the addition of non-message sequences which permit errors in the message sequence to be detected and corrected. It is proposed that sequences in introns (or in other non-message genomic regions) serve this function with respect to the transmission of genetic information. |
|
In eukaryotes the sequence of DNA bases coding for a protein is often found to be interrupted by sequences of bases (introns) which show no obvious relationship to the coding sequence (Gilbert, 1978). Speculation on the possible role of introns has included the view that they are examples of "junk" or "selfish" DNA, which does not contribute positively to cell function (Doolittle & Sapienza, 1980; Orgel & Crick, 1980). However, the notion of message sequences interrupted by non-message sequences is quite familiar to those working on noise affecting signal transmission in electrical systems. In these systems the non-message sequences have an error-checking function and permit the receiver to detect and correct errors in the message sequence (Hamming, 1980). Some principles which may guide investigations of a possible error-checking role for introns are outlined in this paper. 2. Error-correction In-parallel The linear sequence of DNA bases consists of the purines (R, adenine and guanine) and the pyrimidines (Y, thymine and cytosine). When suggesting the duplex structure of DNA, Watson & Crick (1953) pointed out that the sequence of one strand could be derived from the parallel strand, given the algorithm that purines pair only with pyrimidines (adenine with thymine and guanine with cytosine). Thus, a duplex with correct base-pairing can be written: |
RRYRYRRYY |
| If there were "noise" due to an abnormal base (Z ) in one strand of the duplex, then the error could be corrected using information provided by the parallel complementary strand. |
RRZRYRRYY Remove abnormal base RR RYRRYY Insert correct base RRYRYRRYY |
| In this example the error-checking system recognizes Z as abnormal so that it is excised. The correct base is then inserted. However, if the noise were due to a normal base in the wrong position, then the error-checking system would not know which strand contained the correct sequence. There would only be a 50% probability of correcting the error. |
RRRRYRRYY Replace base in top or bottom strand, resulting in either a corrected sequence RRYRYRRYY or a mutation RRRRYRRYY
|
| In this example the error-checking system recognizes that R-with-R pairing is incorrect and changes either the R in the top strand (error corrected) or the R in the bottom strand (error compounded). In a diploid organism containing two homologous parallel copies of duplex DNA it should be possible to correct noise due to a normal base in the wrong position with 100% probability of success. |
| Duplex with error RRRRYRRYY + Homologous duplex RRYRYRRYY Results in: RRYRYRRYY + RRYRYRRYY |
| In this example the error-checking system determines which of the two strands contains the error by comparing the duplex with the error with the homologous duplex. However, if the error were due to a switch in a base pair so that both strands of the duplex were affected without infringement of the purine-with-pyrimidine pairing rule, then comparison with a homologous duplex would permit only a 50% probability of error correction. |
RRRRYRRYY + RRYRYRRYY Resulting in either: |
|
RRYRYRRYY
+ RRYRYRRYY |
RRRRYRRYY + RRRRYRRYY |
| In this example, the error-checking system determines that there has been an error and at what part of the sequence it has occurred. The error-checking system cannot determine which of the homologous duplexes contains the primary error. A solution to the problem would be to bring in further homologous duplexes from other cells (e.g. by sexual mating or by fusion of somatic cells). The extraneous duplexes could then be compared with the cell's own duplexes. |
|
Another solution would be to have homologous duplexes in-series in the cell's own DNA as suggested by Callan (1967) in his "master-slave gene" hypothesis. Callan assumed that the in-series sequences would be identical and that the continuity of the sequence coding for a particular protein would not be interrupted. An explanation of introns in these terms needs to explain why introns interrupt message sequences and why the sequence of an intron is so different from the exons surrounding it? The former question can be answered by considering the structure of information systems. A system of information, such as the paragraph you are now reading, has a structure which imposes itself upon the message sequence. Thus a sentence is interrupted at the end of a line and continues on the next line. It is possible that the proposed error-checking system is structurally constrained so that error-checking sequences must appear in certain positions. This structure would also determine which parts of the DNA sequence are checked by a particular error-checking sequence. Thus the exons next to a particular intron need not be checked by that intron. This could explain why intron sequences are so different from neighbouring exons. Another explanation would be that the algorithm relating intron sequences to exon sequences is different from the classical base-pairing algorithm (Watson & Crick, 1953). An example can be derived by analogy with error-checking codes in electrical systems. One of the simplest error-correcting codes uses a parity check algorithm to detect error (Hamming, 1980). Thus, if for purine we score 1 and for pyrimidine we score zero, a triplet of three bases can be summed with a fourth checking base in order to maintain even parity. |
| Message bases RRY |
Check base Y |
Message bases RYR |
Check base Y |
Message bases RYY |
Check base R |
| The check bases could be collected together and the error-checking system would be able to apply each of them to the appropriate part of the message sequence. |
RRYRYRRYY |
YYR |
|
Detection of non-parity would show that one of the four bases (three message and one check base) was incorrect. To localize the error more precisely, the message could be spread in a two-dimensional matrix (say over the surface of a nucleosome) and two sets of checking bases could be applied. |
RRY Y RRR |
| The message sequence is shown here coiled up and the two checking sequences are arranged vertically and horizontally. There is no requirement for colinearity between message sequences and checking sequences. An error, say in the central of the nine message bases (Y to R), would register with the central base of each checking triplet and so would be precisely located. The ratio of checking bases to message bases would be 2/3. |
|
A system similar to this could exist in eukaryotes for preventing purine-pyrimidine interchanges (transversions), the consequences of which are likely to be more detrimental than purine-purine and pyrimidine-pyrimidine interchanges (transitions). However, eukaryotes have probably evolved a more complex system capable of correcting both transversions and transitions. It is not implausible that DNA molecules could be arranged in cells to produce geometrical arrangements of bases which could be read against checking sequences. Enzymes exist which can literally tie DNA into knots (Cozzarelli, 1980; Liu, Liu & Alberts, 1980). The function of many DNA-binding proteins is unknown, but at least some of these are involved in error-recognition and correction (Kornberg, 1980). There are a number of differences between prokaryotes and eukaryotes in the organization of DNA which would be explained if the latter had evolved a superior error-checking system associated with nucleosomes. Prokaryote DNA polymerases have the ability to "proof-read", yet most eukaryotic DNA polymerases do not have this ability (Kornberg, 1980;[More are now known to have this property]). Prokaryotes do not have a variety of histones, which are the major protein components of nucleosomes (Hubscher, Lutz & Kornberg, 1980). The histones form a core and DNA is wrapped around the core (Laskey & Earnshaw, 1980). According to the geometry of the nucleosome, the winding of the DNA should allow in-series DNA sequences to approach each other, thus providing an opportunity for an error-checking system to operate. In the transversion-detecting model discussed above, a set of four bases would be read in relationship to each other to check that they form one of a subset of acceptable base patterns. Mutations occurring in either checking bases or message bases would be recognized and then, if possible, corrected. Fixation of a mutation in the genome would normally require a concomitant change in two bases of a set. Thus error-checking and message bases would coevolve. Information in introns (or other non-message, error-checking sequences) would coevolve with information in exons. Since the model does not require that an exon be checked by contiguous introns, the latter might evolve at a different rate from surrounding exons (Konkel, Maizel & Leder, 1979). The complex of an exon and its checking intron(s), perhaps located in widely separate parts of the genome, would comprise a unit, permanent damage to any part of which would damage the exon. These information units could be large in organisms with large amounts of DNA and each unit would constitute a large target for mutagenic agents. Thus it would be expected that the mutation rate per locus would increase as the ratio of total genomic DNA to exon DNA increases. Studies of mutagenesis by X-rays and ethyl methane sulphonate are consistent with this (Heddle & Athanasiou, 1975). This paper has presented some principles to assist studies of a possible error-correcting role of introns. It should not be difficult to search for algorithms relating intron sequences to exon sequences. A central computer bank of DNA sequences can be readily accessed (Dayhoff et al., 1980; Gingeras & Roberts, 1980). Rapid computerized decoding techniques have been developed for military purposes. The discovery of such algorithms would justify a search for mechanisms by which exon sequences could be checked against intron sequences. The author thanks Dr Keith Randle of the Royal Military College of Science, Shrivenham, for drawing attention to error-correcting codes, and Dr Stafford Tavares of Queen's University, for helpful discussion. |
|
REFERENCES CALLAN, H. G. (1967). J. Cell Sci. 2, 1. 2, 1. COZZARELLI, N. R. (1980). Science 207, 953. 207, 953. DAYHOFF, M. O., SCHWARTZ, R. M., CHEN, H. R., HUNT, L. T., BARKER, W. C. & ORCUTT, B. C. (1980). Science 209, 1182. 209, 1182. DOOLITTLE, W. F. & SAPIENZA, C. (1980). Nature, Lond. 284, 601. 284, 601. GILBERT, W. (1978). Nature, Lond. 271, 501. 271, 501. GINGERAS, T. R. & ROBERTS, R. J. (1980). Science 209, 1322. 209, 1322. HAMMING, R. W. (1980). In: Coding and Information Theory. pp. 21-49. Englewood Cliffs: Prentice-Hall. pp. 21-49. Englewood Cliffs: Prentice-Hall. HEDDLE, J. A. & ATHANASIOU, K. (1975). Nature, Lond. 258, 359. 258, 359. HUBSCHER, U., LUTZ, H. & KORNBERG, A. (1980). Proc. natn. Acad. Sci. U.S.A. 77, 5097. 77, 5097. KONKEL, D. A., MAIZEL, J. V. & LEDER, P. (1979). Cell 18, 865. 18, 865. KORNBERG, A. (1980). In: DNA Replication. pp. 101-229. San Francisco: Freeman & Co. pp. 101-229. San Francisco: Freeman & Co. LASKEY, R. A. & EARNSHAW, W. C. (1980). Nature, Lond. 286, 763. 286, 763. LIU, L. F., LIU, C. C. & ALBERTS, B. M. (1980). Cell 19, 697. 19, 697. ORGEL, L. E. & CRICK, F. H. C. (1980). Nature, Lond. 284, 604. 284, 604. WATSON, J. D. & CRICK, F. H. C. (1953). Nature, Lond. 171, 263 171, 263 |
The conundrum outlined above was that of determining which was the "sick" strand and which was the "healthy" strand, given knowledge that sickness was present at a duplex site. The proposed solution (1981) envisaged the comparison and correction of homologous duplexes without epigenetic involvement:
Virgil Reese of Bartlesville, Oklahoma, suggested (2002) that the process might best extend transgenerationally if a site were marked as suspicious in one generation so it could be identified in the next generation. Thus, the first comparison within a potential parent organism at meiosis would involve recognition of the problem and site-tagging. In the child there would be a second comparison with a new duplex provided by the other parent. Provided the other parent did not have a tag (mark) at the same site (unlikely), the probability would be that the error was in the tagged site, which would be corrected at meiosis to be like the non-tagged site. If this system could have evolved (i.e. if the hand of evolution could have looked ahead and done something "for the good of" the next generation), then this would provide a nice explanation for why, in some cases, epigenetic modifications are retained between generations (instead of the usual erasure at some point in the reproductive cycle). Sometimes this Lamarckian-style inheritance can have phenotypic consequences other than error-correction (Pembrey et al. 2006). Now we need to work out how such a system might have evolved! Perhaps initially there was a "spandrel" which was later co-opted for another need? Pembrey ME et al. (2006) Sex-specific, male-line transgenerational responses in humans. European Journal of Human Genetics 14, 159-166. Reese V (2002) Mutation repair: a proposed mechanism that would enable complex genomes to better resist mutational entropy and which suggests a novel function for meiosis. Abstracts of Human Behavior and Evolution Society 14th Annual Meeting, Rutgers University. Session on "New Developments in Biology" June 21, p. 40. Click Here |
![]()
Introns, Antirecombination and GC%
| A Buffer Against
Mispairing?
In 1985 when considering gene duplication, Russell F. Doolittle pondered (Trends in Biochem. Sci. June):
Doolittle's focus here is on the prevention of pairing between the sequences of duplicate genes. By preventing pairing, potential recombination events would be discouraged. Since duplicate genes have similar sequences, and recombination can occur when sequences are similar, then the introduction of introns that are more able to accept mutations (i.e. to diversify), would serve to preserve the duplicate genes (i.e. prevent them blending by recombination). This theme was extended to involve differences in GC% and isochores by Matsuo and coworkers, and quotations from their important 1994 paper are here reproduced with the copyright permission of Walter Schaffner. |
Role
of GC%? Introns are usually more highly diversified than neighbouring exons. From detailed studies of POU domain gene families, in a 1994 paper Matsuo and coworkers were led to suggest that "diverged introns would reduce the length of sequence that is common in two DNA segments, resulting in a dramatic reduction of the efficiency of homologous recombination." Thus, the diversification would serve to decrease intragenomic recombination between similar genes. "This would help to explain a long-known paradox, namely that most introns are preserved [between species] even though their actual sequence hardly seems to matter." See paper entitled:
The
authors extrapolated this principle to exons, where third codon
positions, generally free from protein-encoding constraints, can also
play a role in antirecombination:
This suggests a discordance with the general theme of this web-page, which relates to nucleic acid structure. However, structure is exquisitely sensitive to small GC% differences (Click Here Forsdyke, 1998). Thus, it is proposed that GC% differences cause structural differences which, in turn, impede recombination (see below). |
![]()
| Alternative
introns early theory
Letter to the Editor. [Declined circa Nov 1994 by Nature (FXB024A). Published by the author on Bionet 23-08-1995] SIR - In his New & Views item entitled "The uncertain origin of introns"1 Laurence Hurst presents some of the arguments for "introns early" (the Gilbert school 2) and "introns late" (the Stoltzfus school 3). Both schools seem not to have noticed that introns interrupt both coding and non-coding parts of genes4. It has long been known that genes for rRNAs and tRNAs contain interruptions, but these may be special cases. Recently, however, "mRNAs" have been discovered which have no protein product. The corresponding genes look like most protein-encoding genes, and possess multiple introns5. Thus, introns interrupt genetic information, not just protein-encoding information. It is not too surprising then, that it is difficult to associate exons with domains of protein structure or function2,3. It does not follow that this disposes of the introns early viewpoint. There may other exon theories of genes, as well as "the" exon theory of genes. Evidence for an alternative exon theory of genes has come with the discovery that there has been a genome-wide pressure on sequences to accept mutations favouring stem-loop formation6. Adapting the procedure of Le and Maizel7, long sequences from a variety of species were divided into 200 nt windows, each of which was submitted to a computer program which determines the minimum free energy of folding into stem-loop structures. The sequence in each window was then shuffled and again subjected to the folding program. It was found consistently that the folding of natural sequences is energetically more favourable than the folding of the corresponding shuffled sequences. This finding that stem-loop-forming potential is widely dispersed in genomes is consistent with growing evidence that the "kissing" between loops at the tips of stems on homologous chromosomes is rate-limiting in recombination8,9. Mutations which favour recombination may affect either the enzymes involved in recombination, or their substrate, DNA itself (hence stem-loops). In the early "RNA world"10, it is likely that exchange of segments between the prototypic replicators would have been advantageous11. Thus, if it were possible for recombination to have arisen early, it would have done so. The basic postulate of the alternative exon theory of genes is that stem-loop-forming potential was widespread in genomes from an early stage. Information for new functions as they arose had to compete with information for the stem-loop-forming function. In the case of protein-encoding functions the conflict was managed in three ways.
Remarkably, traces of this primitive arrangement can be discerned in some modern genes (D.R.Forsdyke, unpublished work6). In the compact genome of C. elegens stem-loops are abundant and 43% of these occur in introns, which represent only 20% of the genome12. Detailed models for the role of stem-loops in recombination are available13-16. D. R. Forsdyke Department of Biochemistry, Queen's University, Kingston, Ontario, Canada K7L3N6 1. Hurst,L.D. Nature 371, 381-382 (1994). 2. Gilbert,W. & Glynias,M. Gene 135, 137-144 (1994). 3. Stoltzfus et al. Science 265, 202-207 (1994). 4. Hawkins, J.D. Nucleic Acids Res. 16, 9853-9905 (1988). 5. Pfeifer, K. & Tilghman, S.M. Genes Devel. 8, 1867-1874 (1994). 6. Forsdyke, D.R. FASEB.J. 8, 1395A (1994). 7. Le, S-Y. & Maizel, J.V. J. Theor. Biol. 138, 495-510 (1989). 8. Kleckner, N., Padmore, R. & Bishop, D.K. Cold Spring Harbour Symp. Quant. Biol. 56, 729-743. 9. Kleckner, N. & Weiner, B.M. Cold Spring Harbour Symp. Quant. Biol. 58, 553-565. 10. Joyce, G. F. & Orgel, L. E. The RNA World, 1-25 (Cold Spring Harbour Laboratory Press, New York). 11. Bernstein, C. & Bernstein, H. Aging, Sex and DNA Repair, (Academic Press, San Diego). 12. Wilson et al. Nature 368, 32-38 (1994). 13. Sobell, H.M. Proc. Natl. Acad. Sci. USA 69, 2483-2487 (1972). 14. Wagner, R.E. & Radman, M. Proc. Natl. Acad. Sci. USA 72, 3619-3622 (1975). 15. Doyle, G.G. J. Theor. Biol 70, 171-184. 16. Reed et al. J. Mol. Evol. 38, 352-362. |
![]()
A Stem-Loop "Kissing" Model for the Initiation of Recombination and the Origin of Introns
1. Abstract 1994 2. Paper 1995
1. Abstract.
FASEB J.
8, A1395
Washington meeting of Fed. Am. Biol. Socs. Exp. Biol. May 1994
(submission deadline Jan 11 1994) |
2. Paper D. R. Forsdyke (1995a) Molecular Biology and Evolution 12, 949-958.
[JAN KLEIN, reviewing editor; Received February 18, 1995; Accepted May 3, 1995. Reproduced with copyright permission from the Editor, Simon Easteal]
Key words: recombination, stem loop, homology search, introns early, (G+C)/(A+T) ratio, speciation.
| [Technical note (2005) not in the original text: The base order-dependent component of the stem-loop potential of a nucleic acid segment (FORS-D) is derived by a subtraction between the base composition-dependent component (FORS-M) and the total stem-loop potential (FONS). In early papers like this one, the direction of the subtraction was such that FORS-D values were positive when the base order-dependent component was high. However, it is more consistent with the literature to make FORS-D values negative when base order-dependent stem-loop potential is high, and this is adopted in later papers (1998 onwards). In this 1995 paper, a statement such as "FORS-D values correlate negatively with exon overlap" means that, as a DNA segment of fixed length ("window") increasingly overlaps an exon, its base order-dependent stem-loop potential progressively declines. In other words, base order-dependent stem-loop potential is higher in introns.] |
Positive FORS-D Values Are Dispersed in Genomic DNA
The Competing Needs of the Early Replicators
Evidence for a Stem-Loop Model
Conflict Hypothesis and the Evolution of Introns
Why Are FORS-D Values So Sensitive?
Summary. Mutations which improve the efficiency of recombination should affect either the proteins which mediate recombination or their substrate, DNA itself. The former mutations would be localized to a few sites. The latter would be dispersed. Studies of hybridization between RNA molecules have suggested that recombination may be initiated by a homology search involving the "kissing" of the tips of stem loops. This predicts that, in the absence of other constraints, mutations which assist the formation of stem loops would be favored. From comparisons of the folding of normal and shuffled DNA sequences, I present evidence for an evolutionary selection pressure to distribute stem loops generally throughout genomes. I propose that this early pressure came into conflict with later local pressures to impose information concerning specific function. The conflict was accommodated by permitting sections of DNA concerned with a specific function to evolve in dispersed segments. Traces of the conflict seem to be present in some modern intron-containing genes. Thus, introns may have allowed the interspersing of selectively advantageous stem loops in coding regions of DNA. |
|
Introduction
The possession of efficient mechanisms for homologous recombination would appear to be evolutionarily advantageous (Bernstein and Bernstein 1991). Thus, mutations in DNA which improve the efficiency either of the proteins concerned with recombination, or of the DNA to act as a target for these proteins, would be evolutionarily advantageous. Mutations improving the efficiency of proteins would be expected to be localized to the region of the corresponding genes. However, mutations improving the ability of DNA to act as a recombination substrate would be expected to be widely distributed. The view that prior formation of a synaptonemal complex is essential for the initiation of meiotic recombination has recently lost ground to the view that an initial sequence-based homology search precedes chromosome pairing (Hawley and Arbel 1993). In 1971 Crick advanced the "unpairing postulate" that supercoiled duplex DNA in chromatin would form double-stranded stem-loop structures. At the tips of these structures individual strands would unpair, providing the opportunity for interactions with single-stranded DNA at the tips of similar stem loops on other chromosomes. This would allow a homology search and the pairing of homologous chromosomes during meiosis. There was no clear prediction that the primary sequence could have evolved to facilitate this.
|
| Subsequent studies of the mechanism of hybridization between the sense and antisense RNAs involved in regulation of the replication of plasmid ColE I indicated that an initial homology search between the complementary single-stranded RNAs involves weak, reversible, transient "kissing" interactions between the tips of stem-loop structures (Tomizawa 1984). Supporting evidence for a role of stem loops in recombination between RNA genomes was provided by Romanova et al. (1986). This raised the possibility that similar stem-loop structures, generated when duplex DNA adopts a cruciform configuration, are involved in the initial homology search during meiosis (Sobell 1972; Wagner and Radman 1975; Kleckner and Weiner 1993). |
|
|
If stem-loop formation in duplex DNA facilitates the initiation of homologous recombination, then there should have been a mutational pressure to produce dispersed sets of complementary oligonucleotide pairs. It is known that the frequencies of members of complementary oligonucleotide pairs are similar in DNA (Pradhu 1993; Forsdyke 1995b). However, it is not known whether the complementary pairs are sufficiently colocalized to permit extensive stem-loop formation. An algorithm for the computation of "statistically significant" stem-loop potential was introduced by Le and Maizel (1989). This involved comparison of the folding into stem-loop structures of small windows in nucleic acid sequences, with the folding of the same windows after randomizing base order. In this paper, their approach is adapted for the determination of the distribution of stem loops in long DNA molecules. I first examine the distribution of stem-loop potential in some of the long DNA sequences (e.g., 68 kb) which have recently become available. The results support the single-stranded DNA stem-loop "kissing" model. I then speculate that an early genome-wide evolutionary pressure for the formation of dispersed stem-loop structures came into conflict with later local pressures to encode specific functions affecting phenotype. Finally, I present a survey of a variety of modern genes to see if traces of the conflict are still discernible. The results cast a new light on both the exon/intron problem (Gilbert and Glynias 1993; Stoltzfus et al. 1994) and the problem of why different genomes or genome compartments have distinct (G+C)/ (A+T) ratios (Wyatt 1952; Bernardi 1989). The latter is discussed more fully elsewhere (unpublished data; [see Forsdyke 1996a]). |
|
The secondary structure of DNA in single-stranded form is very sensitive to small changes in sequence (Orita et al. 1989;[see footnote]). Provided the length is not excessive, any such sequence can be analyzed using computer programs such as FOLD (Zuker 1989) to arrive at a theoretical optimum secondary structure of minimum free energy. Because of the greater strength of GC bonds relative to AT bonds, a GC-rich sequence tends to have a more stable structure than an AT-rich sequence of the same length. However, it does not follow that the most stable structures are likely to be of most local functional relevance. The bases in a segment might show poor complementarity, but if the few complementary pairs were GC pairs, the stability of the folded molecule might be quite high. Base composition is a genome, or genome sector (isochore), "strategy," which has a major influence on codon choice, particularly of synonymous codons (Grantham et al. 1980). Base composition is not a local "strategy." Codons are a local "strategy." Confidence
that a given secondary structure is of local functional relevance is greater if it can be
shown that the sequence has accepted mutations which enhance stem-loop formation (i.e.,
that the actual sequence of bases, in addition to base composition, has contributed to
secondary structure stability). In principle, this should be possible by comparing the
folding of a natural sequence with that of randomized versions of the same sequence. |
| A natural sequence is but one member of a large set of possible sequences with the same base composition. The average characteristics of this set can be arrived at by randomizing the natural sequence. Randomization (shuffling) destroys information present in the primary sequence (base order), without changing base composition or sequence length. Thus, provided length is kept constant, average characteristics reflect base composition alone. |
|
For each 200-nt window, FOLD first determines the minimum free energy value for folding of the natural sequence (FONS value). This is a function of both base composition and base order and measures the "total stem-loop potential" of a region. Then 10 random sequences are generated from the same window, and each randomized sequence is submitted to FOLD. The mean minimum free energy value for the 10 sequences (FORS-M value) provides a measure of the contribution of base composition alone to the stem-loop potential ("base composition-determined stem-loop potential"). Since the FONS value is usually more negative than the FORS-M value, the difference between the two values (FORS-M less FONS) is usually positive. This
difference (the "FORS-D" value) provides a measure of the contribution of base
order alone to the stem-loop
potential. Thus, a positive FORS-D value defines and quantitates the "base order-determined stem-loop potential." This closely
corresponds to the "segment score" of Le and
Maizel (1989), which is used to assess "statistically
significant" stem-loop potential.
A negative
FORS-D value in a region may mean that base order has been adapted to serve some other
potential. |
|
|
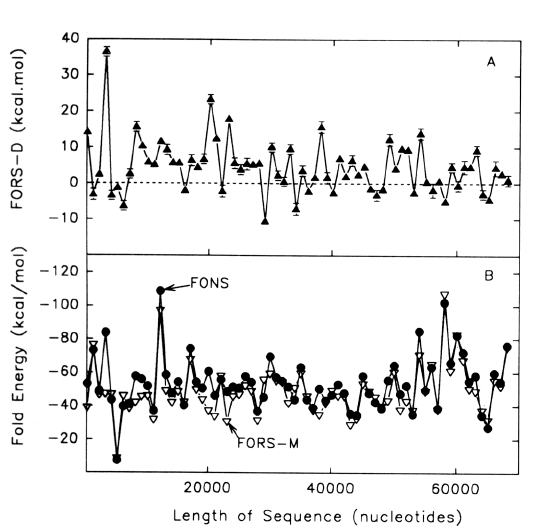
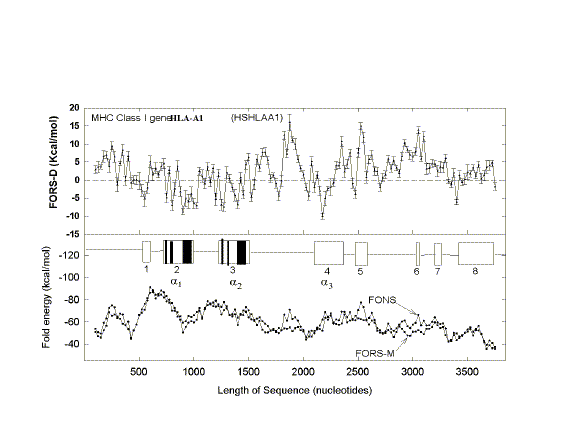
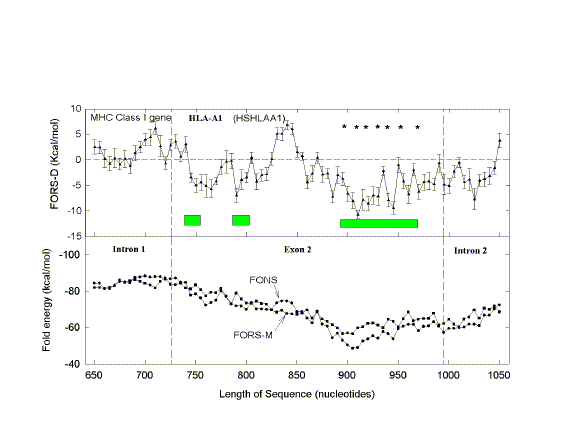
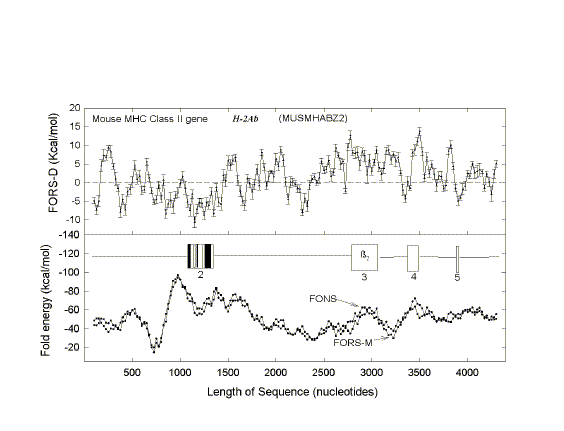
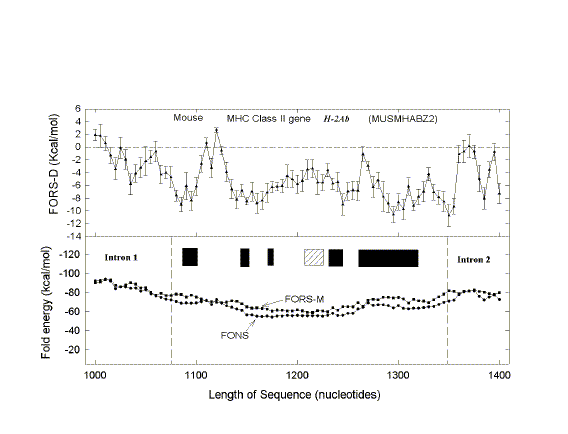
| FIG. 1.- Fold energy minimization values (FORS-M, FONS) and differences (FORS-D) for the 68-kb GenBank sequence HUMMMDBC. Nonoverlapping windows (200 nt) were selected at 1,000-nt intervals (i.e., 1-200, 1,001-1,200, 2,001-2,200, etc.). Secondary structure energy minimization values, determined using the program FOLD, were obtained for each window in the natural sequence (FONS values). Each 200-nt sequence was then subjected to 10 independent randomizations, and FOLD values for each of the 10 randomized sequences (FORS) were determined. In B the mean FOLD value for each set of 10 randomized sequences (FORS-M) is plotted with the corresponding FONS value. In A the differences between the FORS-M values and the corresponding FONS values are plotted (FORS-D values + standard error). FORS-M and FONS values are both negative. FORS-D values are positive whenever the FONS value is more negative (i.e., corresponds to a higher folding energy) than the FORS-M value. Each data point is at the middle of its 200-nt window. |
|
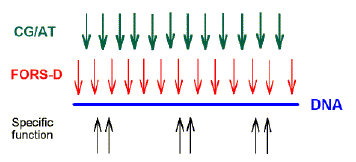
Positive FORS-D Values Are Dispersed in Genomic DNA A study of folding energies was carried out on one strand of the 68-kb human chromosome 19 segment HUMMMDBC (GenBank name). If there had been a genome-wide pressure to maximize the potential to form secondary structures, then positive FORS-D values would be expected. To get a general impression of the folding propensity, the FOLD program was applied to isolated 200-nt windows, at 1-kb intervals. The FONS and FORS-M profiles tend to follow each other (fig. I B). Positive FORS-D values are widely dispersed and greatly exceed negative FORS-D values (fig. 1A; average 4.37+0.90 kcal/mol). Similar results are obtained for the HUMHBB segment from chromosome 11 (average 4.49+1.34 kcal/mol for 15 windows at 5-kb intervals) and for the HUMHDABC segment from chromosome 4 (average 3.59+1.62 kcal/mol for 12 windows at 5-kb intervals). Thus positive FORS-D values are widely dispersed in segments from small, medium, and large human chromosomes. Long DNA fragments from Drosophila melanogaster, Escherichia coli, and bacteriophage lambda also showed significant positive average FORS-D values (Forsdyke 1995b). The dispersal of positive FORS-D values throughout genomes indicates a general evolutionary pressure on DNA molecules to accept mutations which would promote stem-loop formation by affecting base order. This supports the stem-loop "kissing" model for recombination as outlined in the Introduction. FORS-D profiles show negative values in some regions, suggesting conflicts between a general pressure on a gene to optimize its folding propensity and local pressures for some function. Potential conflicts between general and local pressures on evolving DNA molecules are summarized in figure 2. The two rows of downward-pointing arrows symbolize pressures acting throughout the genome. One of these is the general pressure to set the (C+G)/(A+T) ratio (base composition) at a particular level in a particular genome or genome segment ("CG/AT pressure"; see Discussion). The second is the pressure, quantified as the FORS-D, to generate sequences with complementary oligonucleotides situated so as to favor the formation of stem loops. These two sets of arrows are pointing in the same direction, indicating that there is little conflict between them. The upward-pointing arrows are in distinct regions, symbolizing localized evolutionary pressure for the encoding of specific function. Here there is a conflict. A sequence required to encode a protein might not simultaneously be able to optimize its folding propensity. |
|
FIG. 2 |
|
|
|
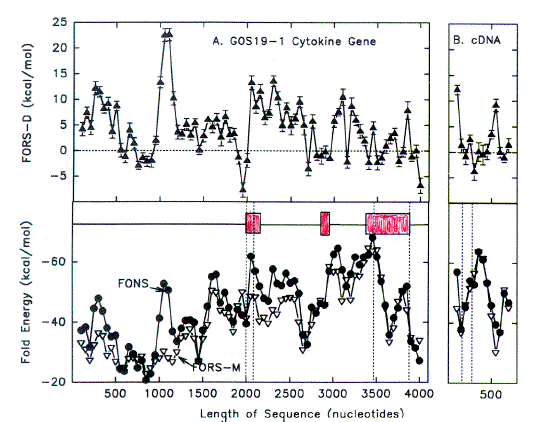
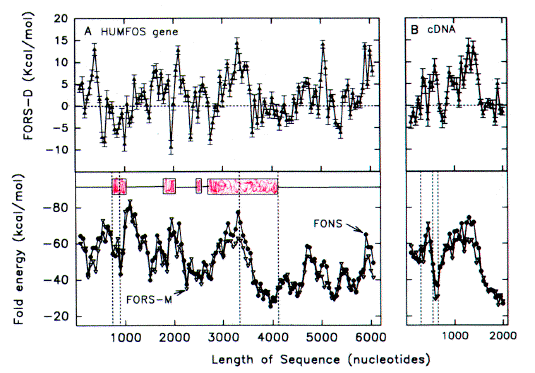
| FIG. 3.-Fold energy minimization values (FORS-M, FONS) and differences (FORS-D) for A, the 4,102-nt sequence containing the G0S19-1 cytokine gene (GenBank name HUMG0S19A), and B, the corresponding cDNA. Each data point corresponds to the middle of a 200-nt window. Each window overlaps the preceding window by 150 nt, except that the first of the 41 overlapping windows of the genomic sequence spans nt 1- 94. Thus windows, in order, correspond to nt 1- 1 94, nt 45-244, nt 95-294, and so forth. This determines that the beginning of exon I corresponds to a new window (nt 1,995-2,194). In B there are 14 overlapping windows which begin with nt 1. The last window of the 778-nt cDNA sequence spans nt 601-778. The three exons are shown as open boxes. Vertical dashed lines in A indicate, from left to right, the beginning of exon 1, the beginning of the protein-encoding region, the end of the protein-coding region, and the end of exon 3. Vertical dashed lines in B show where introns have been removed. |
|
FORS-D Values of Intron-Containing Genes To seek traces of this postulated evolutionary conflict between a genome-wide FORS-D pressure and local pressures related to individual gene function, the folding patterns of some modern genes were examined. Fold energy plots were prepared using consecutive 200-nt windows with 150-nt overlaps. The human cytokine-encoding gene G0S19-1 is one of a series of potential lymphocyte G0/G1 switch regulatory genes ("G0S genes") which have been sequenced in my laboratory (Blum et al. 1990; [Now more widely known as MIP1-alpha or CCL3, whose receptor is a coreceptor for HIV-1]). The gene has three exons distributed over a region of 1,886 nt, and the cDNA derived by splicing is 778 nt in length [See diagram at top of page]. As noted above (fig. 1), FONS and FORS-M profiles tend to resemble each other (fig. 3A, lower panel). FORS-D values are generally positive in the gene; however, distinct local decreases occur in certain regions (fig. 3A, upper panel). When the introns are removed to generate the cDNA, decreased FORS-D values tend to be concentrated together, and general positivity is less evident (fig. 3B, upper panel). An extremely low FORS-D value in the immediate 5'flank of the gene corresponds to a region rich in potential regulatory motifs, which is highly conserved between G0S19-1 and its murine homolog (Russell and Forsdyke 1993).This supports the idea of a conflict between a general evolutionary pressure for stem-loop potential and local pressure for function. The very high positive FORS-D values (20 kcal/mol) in the 5'flank of the G0S19-1 gene correspond to a potentially "foreign" AT-rich minisatellite-like element containing four 22-nt tandem repeats and two inverted repeats. (Only 1 of the 22-nt repeats is present in the G0S19-2 gene. Thus an amplification/contraction probably occurred following the divergence of the two genes; Blum et al. 1990.) Low FORS-D values in the regions of the second and third exons are also consistent with the conflict hypothesis; however, values are high in the signal peptide-encoding first exon (fig. 3A). |
|
|
| FIG. 4.- Fold energy minimization values (FORS-M, FONS) and differences (FORS-D) for the 6,210-nt sequence containing the human c-fos gene (GenBank name HUMFOS). Details are as in fig. 3. |
| Another G0S gene (G0S7) corresponds to the oncogene c-fos, which has four exons (fig. 4). Each exon is associated with a region of negative FORS-D value, whereas the three introns tend to have high FORS-D values. The negative values of the exons are not reflected in the cDNA, which shows no consistent tendency to low values (fig. 4B). The coding part of the fourth exon is long (642 nt). Although the 5' part of the exon is associated with a region of low FORS-D value, the rest of the coding region is associated with high FORS-D values. Thus, here there appears to be no conflict between pressures for the evolution of protein-encoding function and for the potential to form stem loops. The 3' part of the 3' non-coding region has relatively low FORS-D values, consistent with the known functional role of this region in controlling mRNA stability. |
|
|
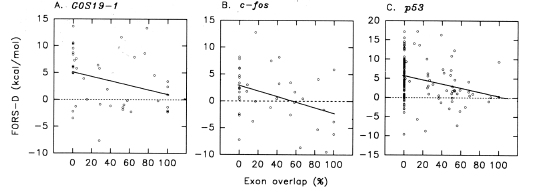
| FIG. 5.- Linear regression analysis of the FORS-D value against the degree of exon overlap(%) for A, G0S19-1 ; B, c-fos; and C, p53. Each data point represents values for a 200-nt window. Each window overlaps its neighbors by 150 nt. For G0S19-1, windows extend from the beginning of the first exon to the end of the last exon. Windows in the flanks are omitted except those which are close to, and hence partially overlap, the first and last exons. For reasons given in the text, terminal exons are omitted from the analysis of c-fos and p53. |
|
FORS-D Values Correlate Negatively with Exon Overlap To obtain an objective measure of the tendency of small exons to associate with low FORS-D values, each of the 200-nt windows for which FORS-D values were determined was scored for its percentage of exon overlap. Figure 5A shows a linear regression plot for the three exons of the G0S19-1 gene. Consecutive windows extended from the first 5'window overlapping the first exon to the last 3'window overlapping the last exon. Although there was much scatter of data points, the slope of the least-squares regression line was significantly greater than zero (P = 0.048 that the slope is not greater than zero). Figure 5B and C show similar data for the oncogenes cfos and p53, except that the long last exons were omitted. Both slopes were significantly greater than zero (P = 0.018, c-fos, P = 0.002, p5 3). The corresponding P values when the last exons were included were 0.752 and 0.048. (A justification for omitting last exons is given later.) |
| GENE | GENBANK NAME |
LENGTH STUDIED (nt) |
EXONS WITH DECREASED FORS-D VALUESc | REGRESSION | ||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | Sloped | P | |||
| Oncogene p53 | HSP53G | 8,750 | . | - | + | - | - | - | + | + | (-) | - | (+) | . | -0.054* | 0.002 |
| Triose phosph. isomerase | HSTPI1G | 4,400 | (-) | - | (+) | (+) | (-) | (+) | (+) | . | . | . | . | . | -0.011 | 0.710 |
| Albumin | HUMALBGC | 8,400 | (-) | (-) | - | - | - | + | . | . | . | . | . | . | -0.026 | 0.232 |
| alpha-B- crystallin |
HUMCRYABA | 4,200 | (-) | (+) | (+) | . | . | . | . | . | . | . | . | . | +0.009 | 0.616 |
| Cytochrome C | HUNCYCAA | 3,088 | - | - | (+) | . | . | . | . | . | . | . | . | . | -0.033* | 0.325 |
| Oncogene c-fos | HUMFOS | 6,210 | - | - | - | (-) | . | . | . | . | . | . | . | . | -0.052* | 0.018 |
| Cytokine G0S19-1 | HUMG0S19A | 4,100 | + | - | (-) | . | . | . | . | . | . | . | . | . | -0.041 | 0.048 |
| Zinc finger protein | HUMG0S24B | 3,135 | - | (+) | . | . | . | . | . | . | . | . | . | . | -0.056* | 0.354 |
| Beta-globin | HUMHBB | 3,100 | (-) | (-) | + | . | . | . | . | . | . | . | . | . | +0.008 | 0.701 |
| Heat shock protein 86A | HUMHSP86A | 2,598 | . | + | - | (+) | - | + | . | . | . | . | . | . | -0.007 | 0.754 |
| Heat shock protein 90B | HUMHSP90B | 8,200 | (+) | - | (-) | + | - | (-) | - | + | - | (+) | - | - | -0.038 | 0.001 |
| Troponin-C | HUMTROC | 4,400 | + | + | - | - | - | + | . | . | . | . | . | . | -0.135 | <0.001 |
| Tcp-10 | MUSTCP10AA | 3,882 | . | - | - | (+) | + | + | + | - | (-) | + | - | . | -0.066 | 0.017 |
| Glucose reg. protein | TOMBIPGRP | 2,966 | (-) | (+) | + | (+) | (+) | . | . | . | . | . | . | . | +0.017 | 0.228 |
| aExons
showing relative decreases in FORS-D values (exemplified by Figs. 3-4) are indicted by
minus signs (-). Exons failing to show decreases in FORS-D values are
indicated by plus signs (+). Parentheses around signs indicate some
ambiguity. bFORS-D values were plotted against degree of exon overlap (exemplified by Fig. 5). P values indicate the extent to which slopes are not significantly different from zero. cAll exons were studied except HUMALBGC (first six only), and HSP53G, HUMHSP86A, and MUSTCP10AA (not first exon. dValues with asterisks indicate that the last exon was omitted from the analysis. |
||||||||||||||||
| The above analyses were extended to a broader spectrum of genes, and the results are shown in table 1. Five of 12 human genes had significant P values (< 0.05 that the slope is not significantly greater than zero). The one mouse gene studied had a significant P value. The one plant gene studied (TOMBIPGRP) did not have a significant P value. The most significant P value was obtained with a gene (encoding troponin C), believed to have been under positive Darwinian selection (Ohta 1994). Table 2 compares average FORS-D values of gene-containing segments (table 1) with those of the corresponding cDNAs. In most cases average FORS-D values for the genomic segments were greater (average +0.71+0.33; P < 0.05 by Student's t-test). |
| FORS-D (kcal/mol) | |||
| GENE NAME | Gene | cDNA | DIFFERENCE |
| Oncogene p53 | 4.8+0.4 (172) | 4.2+0.7 (47) | +0.6 |
| Triose phosphate isomerase | 3.7+0.6 (85) | 4.5+0.7 (22) | -0.8 |
| Albumin | 2.3+0.4 (165) | 0.4+1.1 (12) | +1.9 |
| alpha-B-crystallin | 3.3+0.5 (81) | 1.8+1.0 (10) | +1.5 |
| Cytochrome C | 1.5+0.5 (59) | 2.2+1.0 (22) | -0.7 |
| Oncogene c-fos | 1.9+0.5 (120) | 3.0+0.8 (39) | -1.1 |
| Cytokine G0S19-1 | 4.2+0.6 (79) | 1.8+1.2 (13) | +2.4 |
| beta-globin | 2.7+0.5 (59) | 1.0+1.2 (10) | +1.7 |
| Heat shock protein 86A | 2.2+0.6 (49) | 2.5+1.1 (17) | -0.3 |
| Heat shock protein 90B | 2.2+0.4 (161) | 0.9+0.7 (48) | +1.1 |
| Troponin-C | 4.4+0.7 (85) | 2.3+1.9 (11) | +2.1 |
| Mouse TCP-10 | 3.4+0.6 (75) | 2.6+1.0 (24) | +0.8 |
| Tomato glucose-regulated protein |
2.3+0.5 (56) | 2.2+0.6 (22) | +0.1 |
| NOTE: The average FORS-D values (+ standard error) for 200 nt windows were calculated for DNA segments containing the genes shown in table 1 and their corresponding cDNAs. Numbers of windows, each overlapping the preceding window by 150 nt, are shown in parenthesis. All genes are human except the bottom two. | |||
|
The Competing Needs of the Early Replicators The winner in the competition between replicator molecules in early evolution would have been the replicator which could most efficiently balance the needs for:
Intimately linked with the latter two needs would be the need to carry out repair of malreplicated or damaged molecules. In an early "RNA world," the ability to shuffle damaged segments between molecules so as to create segment combinations which could continue rapid and accurate replication could have been advantageous. Thus, it can be envisaged that a fourth need,
would have been another property which successful replicators would have optimized at an early stage. If Tomizawa's stem-loop "kissing" model is applicable to the early RNA world, then it can be further envisaged that replicators which modified their sequences to increase the probability of stem-loop formation (and hence of recombination), would have had a survival advantage. To reap the benefits of efficient recombination a replicator would have had to exchange segments with its own kind of replicator, not with other kinds. Thus need 4 implies a fifth need,
Finally, when the competition to build better "survival machines" began, a sixth need arose. This was the need to dedicate parts of the genome to the
This discussion deals with potential conflicts between needs 4 and 6, and needs 4 and 5, and suggests how these conflicts might have been resolved. Evidence for a Stem-Loop Model It can be imagined that as part of the attempt to improve replicator stability (need 3), RNA molecules would have been superseded by DNA molecules, but these would have retained the potential to form stem loops. This leads to the prediction explored here that, where not constrained by other needs, modern DNA molecules would have a special potential to form stemloop structures. This would be particularly evident during meiosis (Kleckner and Weiner 1993) and might require an exceptional degree of supercoiling for DNA to depart from its classical duplex structure (Murchie et al. 1992). Consistent with this, inhibitors of topoisomerases, which relax supercoiled DNA, greatly enhance recombination (Wang et al. 1990). Furthermore, endonucleases involved in recombination, which make single-strand nicks in double-stranded DNA, require a supercoiled substrate (Sung et al. 1993). Although a relatively crude measure, there is evidence from studies with anticruciform antibodies for the presence of stem-loop structures, which can vary with the physiological state of the cell (Ward et al. 1990). Further evidence for a role of stem loops in recombination is presented elsewhere (Romanova et al. 1986; Reed et al. 1994). Positive FORS-D values indicate the existence of an evolutionary pressure on DNA base order, rather than on base composition, to generate the potential to form stem-loop structures. Thus, in the absence of other constraints, base changes which enhance the formation of stem loops would have been accepted. I have reported here that dispersed positive FORS-D values are found in long human genome segments considered to consist mainly of nontranscribed intergenic DNA (fig. 1). This is found with DNA from various species, including some with much less intergenic DNA than humans (Forsdyke 1995b). The most obvious genome-wide selective force driving evolution of the potential to form stem loops is recombination. Conflict Hypothesis and the Evolution of Introns A potential conflict between the genome-wide FORS-D pressure and pressures on DNA to encode specific functions is apparent (fig. 2). A sequence required to encode a protein might not at the same time be able locally to optimize its folding propensity. The conflict can be met in three ways.
If the first two options are not sufficient, then only the third option is left. Thus introns might correspond to parts of a gene where the constraints on the first two options are most severe. The more severe the constraints, the more introns there would be, and the longer would be the length of DNA occupied by the gene. This conflict hypothesis might explain how introns initially arose. However, it does not appear to address the problem of how introns came to vary so dramatically in length (Hawkins 1988). Variation in intron length would tend to prevent recombination between genes with homologous exon sequences by impairing the precise register required for successful "kissing" between the loops of stem-loop structures. In terms of FORS-D values, the hypothesis states that when exons first arose, the exon sequences themselves would have had low FORS-D values, except where, by chance, the need to encode a peptide happened not to be in conflict with the need to form stem loops. The low FORS-D exons would have been surrounded by high FORS-D introns and flanking sequences. Over evolutionary time the demands of regulation of gene expression would have been imposed upon this primitive arrangement, so that low FORS-D values might be present in regions of DNA where regulatory proteins bind (except, perhaps, where the regulatory proteins recognize DNA palindromes). Also, over evolutionary time a background "noise" might have been imposed by mobile genetic elements and recombinations accompanying exon shuffling (Gilbert and Glynias 1993). Taking into account the proposed long evolutionary period since introns were established in genes, the analyses of FORS-D values in various intron-containing genes (figs. 3-4; table 1) do provide some support for the idea of a conflict between genome-wide and local pressures. Further evidence has been provided by recent studies of snake venom phospholipase A2 genes, retroviral genomes, and major histocompatibility complex peptide-binding genes (Forsdyke 1995a, and unpublished data [see below]). All of these are under greater selection pressure (positive selection) than the genes studied here (with the possible exception of genes encoding troponin C; Ohta 1994). Some terminal long open-reading frames were excluded from the linear regression analyses shown in figure 5 and table 1. In terms of the hypothesis presented here, long open-reading frames would only exist in certain genes because those genes had been able to resolve the conflict in the regions where we now see the long open-reading frames; FORS-D pressure would have been accommodated by the choice of appropriate synonymous codons or of amino acids with similar functions. (These would have been the main choices open to organisms without introns.) Thus, FORS-D analyses of genes with long open-reading frames are not inconsistent with the conflict hypothesis (fig. 4). By omitting unduly long exons from regression analyses, one hopes to better assess genomic regions which are less able to adapt to FORS-D pressure by using synonymous codons or functionally similar amino acids. The conflict hypothesis would predict that cDNA sequences would tend to have lower average FORS-D values than the genes from which they derive. However, it does not follow that low FORS-D exons, when united, will always result in a low FORS-D cDNA. Two independently arising exons may each have a low potential to form stem loops but a high potential to form stem loops collectively. As a simple example, consider two exons of sequence CACACACA and TGTGTGTG. Both have low FORS-D values, but they would unite to generate a cDNA with a very stable stem loop (high FORS-D value). Only some genes show clear evidence for higher average FORS-D values than the corresponding cDNAs (G0SI 9-1, fig. 3; table 2). Why Are FORS-D Values So Sensitive? Evolutionary pressures (mutation, selection, drift) determine how many bases there are in a sequence (DNA length), the proportions of different bases (base composition), and the order of the bases (sequence). The experimental shuffling of the order of bases in a sequence of given length and base composition disrupts information present in the natural sequence and generates a reference sequence of the same length and base composition against which the natural sequence can be compared. Thus one can distinguish evolutionary pressures affecting base order (primary sequence) from those affecting base composition. FORS-M and FONS profiles closely follow each other (figs. I B, 3B-4B), showing that base composition is the major determinant of the stability of stem loops. It thus might seem somewhat surprising that differences of only a few kilocalories (FORS-D values) provide such a sensitive indicator. That fluctuations in FORS-D values are not random is shown by their consistent average positivity (fig. 1) and the association of negative FORS-D values with functionally important regions of DNA (fig. 3). Base composition is a genome "strategy" (Grantham et al. 1980) and thus is less likely to be relevant to local gene function than primary sequence. FORS-D values provide a sensitive indicator because they measure the base order-determined stem-loop potential, not the base composition-determined stem-loop potential. A close examination of folding energy profiles reveals that high FORS-D values may sometimes be associated with quite low negative FONS values (a function of base order and base composition) and even lower negative FORSM values (a function of base composition). Thus, high negative FONS values (implying a very stable secondary structure) may sometimes have little local functional relevance. The recombination model of Wagner and Radman (1975) proposes that, after initial contact is made by loops at the tips of the stems, there are endonucleolytic cleavages of single strands of opposing stems. Single strand exchanges between the stems then follows. This process would presumably abort if precise complementarity were not present in the stems (Radman and Wagner 1993). Furthermore, Tomizawa (1993) has concluded that the major role of the stem in a stem-loop structure is the proper positioning of the loop. This allows the unpaired bases in the loop to pair, in register, with those of an appropriately positioned loop projecting from a complementary nucleic acid. This "kissing" is rate limiting in recombination. Thus, a departure, even by a few kilocalories, in complementarity between bases in the stem might prevent recombination. An evolutionary force adapting base order to favor stem-loop formation could be a very powerful one. In that the ability to recombine is held to have evolved before the ability to encode proteins, this paper supports the "introns early" model. The positions of exon-intron junctions are held to have been determined by the need to form stem loops and are not necessarily related to protein domains (Gilbert and Glynias 1993). Stoltzfus et al. (1994, p. 202) concluded that "no significant correspondence between exons and units of protein structure was detected." GC/AT Fine-Tuning and Speciation Different genomes, or genome compartments, have characteristic (G+C)/(A+T) ratios (Wyatt 1952; Bernardi 1989; Filipski 1990). In vertebrates this "GC/AT pressure" (fig. 2) is evident in intergenic regions, and introns, and in both coding and noncoding parts of exons. The "genome hypothesis" (Grantham et al. 1980) has led to the proposal that there is some intrinsic genome-wide pressure favoring the adoption of a particular (G+C)/(A+T) ratio in a particular species. The view that this primarily reflects mutational biases (Filipski 1990) has been challenged (Bernardi 1989). I argue elsewhere (unpublished data), that species-specific settings of the (G+C)/(A+T) ratio have arisen as part of a fine-tuning process which prevents recombination between species. Just as different radio transmitters broadcast their messages at different wavelengths to avoid interference, so different genomes "broadcast" their sequences at different (G+C)/(A+T) "wavelengths," to avoid undesirable recombination events. The present work suggests a mechanism by which this could occur. Base composition, rather than base order, is the major factor determining the folding energy of a nucleic acid segment (figs. 1B, 3B-4B). Thus small changes in base composition could greatly affect the looping pattern which a sequence could present for homology search. It would be more difficult to extrude loops from GC-rich DNA than from AT-rich DNA.
If avoidance of recombination between different species (including a host species and its pathogen species) is evolutionarily advantageous (Bernstein and Bernstein 1991), then there should have been a selection pressure such that each species in a common environment with other species would have fine-tuned its (C+G)/(A+T) ratio to a level distinct from those of the other species. Thus, biologically similar species may have dissimilar ratios, whereas biologically dissimilar species (unlikely to interact sexually and with dissimilar pathogens) may have similar ratios (Wyatt 1952). The fine-tuning of base ratios could have been a key component of the postzygotic isolation process leading to speciation (unpublished data; [see Evolution Web Page]). Acknowledgments I thank D. Back for assistance in computer configuration and M. Go, C. Tittiger, V. K. Walker, and G. R. Wyatt for helpful comments on the manuscript. The work was supported by a grant from the Medical Research Council of Canada. |
|
LITERATURE CITED BERNARDI, G. 1989. The isochore organization of the human genome. Annu. Rev. Genet. 23:637-661. BERNSTEIN, C., and H. BERNSTEIN. 199 1. Aging, sex and DNA repair. Academic Press, San Diego. BLUM, S., R. E. FORSDYKE, and D. R. FORSDYKE. 1990. Three human homologs of a murine gene encoding an inhibitor of stem cell proliferation. DNA Cell. Biol. 9:589-602. CRICK, F. 197 1. General model for the chromosomes of higher organisms. Nature 234:25-27. FILIPSKI, J. 1990. Evolution of DNA sequence: contributions of mutational bias and selection to the origin of chromosomal compartments. Adv. Mutagenesis Res. 2:1-54. FORSDYKE, D. R. 1995a. Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes: an application of FORS-D analysis. Mol. Biol. Evol. 12:1157-1165. FORSDYKE, D. R. 1995b. Relative roles of primary sequence and (G+C)% in determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J. Mol. Evol. 41:573-581. GILBERT, W., and M. GLYNIAS. 1993. On the ancient nature of introns. Gene 135:137-144. GRANTHAM, R., G. GAUTIER, M. GOUY, R. MERCIER, and A. PARE. 1980. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 8: r49-r62. GRIBSKOV, M., and J. DEVEREUX. 1991. Sequence analysis primer. Stockton, New York. HAWKINS, J. D. 1988. A survey of intron and exon lengths.Nucleic Acids Res. 16:9893-9905. HAWLEY, R. S., and T. ARBEL. 1993. Yeast genetics and the fall of the classical view of meiosis. Cell 72:301-303. KLECKNER, N., and B. M. WEINER. 1993. Potential advantages of unstable interactions for pairing of chromosomes in meiotic, somatic and premeiotic cells. Cold Spring Harbor Symp. Quant. Biol. 58:553-565. LE, S.-Y., and J. V. MAIZEL. 1989. A method for assessing the statistical significance of RNA folding. J. Theor. Biol. 138: 495-510. MURCHIE, A. 1. H., R. B0WATER, F. ABOUL-ELA, and D. M. J. LILLEY. 1992. Helix opening transitions in supercoiled DNA. Biochem. Biophys. Acta 1131:1-15. OHTA, T. 1994. Further examples of gene duplication revealed through DNA sequence comparisons. Genetics 138:1331-1337. ORITA, M., H. IWAHANA, H. KANAZAWA, K. HAYASHI, and T. SEKIYA. 1989. Detection of polymorphisms of human DNA by gel electrophoresis as single strand conformation polymorphisms. Proc. Natl. Acad. Sci. USA 86:2766-2770. PRABHU, V. V. 1993. Symmetry observations in long nucleotide sequences. Nucleic Acids Res. 21:2797-2800. RADMAN, M., and R. WAGNER. 1993. Mismatch recognition in chromosomal interactions and specification. Chromosoma 102:369-373. REED, K. M., L. W. BEUKEBOOM, D. G. EICKBUSH, and J. H. WERREN. 1994. Junction between repetitive DNAs on the PSR chromosome of Nasonia vitripennis.- association of palindromes with recombination. J. Mol. Evol. 38:352-362. ROMANOVA, L. 1., V. M. BLINOV, E. A. TOLSKAYA, E. G. VIKTOROVA, M. S. KOLESNIKOVA, E. A. GUSEVA, and V. 1. AGOL. 1986. The primary structure of crossover regions of intertypic poliovirus recombinants: a model of recombination between RNA genomes. Virology 155:202-213. RUSSELL, L., and D. R. FORSDYKE. 1993. The third human homolog of a murine gene encoding an inhibitor of stem cell proliferation is truncated and linked to a CpG island-containing upstream sequence. DNA Cell. Biol. 12:157-175. RYAN, B. F., and B. L. JOINER. 1994. Minitab handbook. 3d ed. Wadsworth, Belmont, Calif SOBELL, H. M. 1972. Molecular mechanism for genetic recombination. Proc. Natl. Acad. Sci. USA 69:2483-2487. STOLTZFUS, A., D. F. SPENCER, M. ZUKER, J. M. LOGSDON, and W. F. DOOLITTLE. 1994. Testing the exon theory of genes: the evidence from protein structure. Science 265: 202-207. SUNG, P., P. REYNOLDS, L. PRAKASH, and S. PRAKASH. 1993. Purification and characterization of the Saccharomyces cerevisiae RADI/RADIO endonuclease. J. Biol. Chem. 268: 26391-26399. TOMIZAWA, J. 1984. Control of ColE I plasmid replication: the process of binding of RNA I to the primer transcript. Cell 38:861-870. TOMIZAWA, J. 1993. Evolution of functional structures of RNA. Pp. 419-445 in R. F. GESTELAND and J. F. ATKINS, eds. The RNA world. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y. TURNER, D. H., and P. C. BEVILACQUA. 1993. Thermodynamic considerations for evolution by RNA. Pp. 447-464 in R. F. GESTELAND and J. F. ATKINS. eds. The RNA world. Cold Spring Harbor, Cold Spring Harbor, N.Y. TURNER, D. H., N. SUGIMOTO, and S. M. FREIER. 1988. RNA structure prediction. Annu. Rev. Biophys. Chem. 17:167192. WAGNER, R. E., and M. RADMAN. 1975. A mechanism for initiation of genetic recombination. Proc. Natl. Acad. Sci. USA 72:3619-3622. WANG, J. C., P. R. CARON, and R. A. Kim. 1990. The role of DNA topoisomerase in recombination and genome stability: a double-edged sword? Cell 62:403-406. WARD, G. K., R. McKENZIE, M. ZANNIS-HADJOPOULOS, and G. B. PRICE. 1990. The dynamic distribution and quantification of DNA cruciforms in eukaryotic nuclei. Exp. Cell Res. 188:235-246. WYATT, G. R. 1952. The nucleic acids of some insect viruses. J. Gen. Physiol. 36:201-205. ZUKER, M. 1989. Computer prediction of RNA secondary structure. Meth. Enzym. 180: 262-289. |
|
End
note
2001: Citing Orita et al (1989) it is noted above that "The secondary
structure of DNA in single-stranded form is very sensitive to small changes in sequence."
The exquisite sensitivity of secondary structure to a change in even a
single base is emphasized by later papers:
Shen, L. X., Basilion, J. P. & Stanton, V. P. (1999) Single nucleotide polymorphisms can cause different structural folds of mRNA. Proc. Natl. Acad. Sci. USA 96, 7871-6. Dong, F., Allawi, H. T., Anderson, T., Neri, B. P. & Lyamichev, V. I. (2001) Secondary structure prediction and structure-specific sequence analysis of single-stranded DNA. Nucleic Acids Res. 29, 3248-57. |
![]()

Conservation of Stem-Loop Potential in Introns of Snake Venom Phospholipase A2 Genes. An Application of FORS-D Analysis
D. R. Forsdyke (1995b). Molecular Biology and
Evolution 12, 1157-1165.
[JAN KLEIN, reviewing editor; Received
February 16, 1995; Accepted May 19, 1995. With copyright permission from the Editor, Simon
Easteal]
Keywords:
Abbreviations:
|
High substitution density in exons
Reciprocal relationship between substitution density and FORS-D
|
Summary. Venomous snakes and their
prey are engaged in an "arms race" in which resistance to venom proteins must be
countered by the adaptation of the proteins to overcome that resistance. The toxicity of
venoms is contributed to by phospholipase A2 the genes of which are under
extreme positive selection pressure. In contrast to most other gene families, intraspecies
and interspecies comparisons of venom phospholipase A2 genes reveal low
conservation of exons and high conservation of
introns. FORS-D analysis ("folding of randomized sequence difference analysis") implies that the sequences of introns of venom phospholipase A2 genes have been under pressure to accept mutations which conserve the potential to form single-strand stem-loops, whereas exon sequences have evaded this pressure. These results are consistent with the hypothesis that stem-loop-based recombination evolved in the early "RNA world", and the potential to generate stem-loops became widely dispersed throughout genomes. The subsequent conflict between stem-loop potential and protein-encoding potential was resolved by:
In genes under strong Darwinian selection, the pressure on genes to diversify in order to prevent unwanted recombination events is largely accommodated by exons. This leaves introns, which normally predominate in this role, free to accommodate to pressure for stem-loop formation. |
|
In a previous paper (Forsdyke, 1995a) it was suggested that, if efficient recombination is evolutionarily advantageous, then it is possible that it would have evolved in the early "RNA world", prior to the emergence of protein-encoding genes (Joyce & Orgel, 1993). If this were so, and if stem-loops were involved in this primitive recombination process (Romanova et al. 1986; Kleckner et al. 1991; Kleckner and Weiner, 1993), then protein-encoding genes would have been imposed on sequences which had already been adapted for stem-loop formation. There could then have been a conflict, since a sequence might not be capable of locally optimizing both its base order-determined stem-loop potential and its base order-determined protein-encoding potential. The conflict could have been accommodated in three ways:
The latter "introns early" theory stated that the early introns would have been regions of high base order-determined stem-loop potential, and predicted that traces of this primitive arrangement should be detectable in some modern genes. To examine this, FORS-D analysis was applied to various gene sequences. A tendency for base order-determined stem-loop potential to localize to introns was apparent, but in several cases the potential was equally apparent in exons and introns (Forsdyke, 1995a). What was needed was a gene that had been under such extreme selection pressure for function that accommodation of the potential in exons would have been unlikely. |
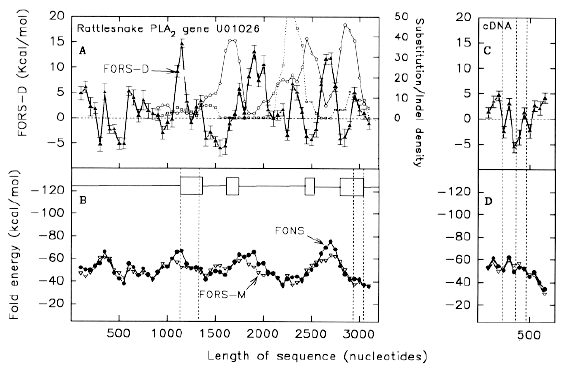
| The snake venom phospholipase A2 (PLA2) genes appear ideal for this purpose. Snakes are engaged in an intense "arms-race" with their prey (or predators), which can acquire venom resistance (Harding and Welch, 1980). Recently, the genomic sequences of PLA2 genes have become available from habu snakes (Nakashima et al. 1993), and rattlesnakes (John et al. 1994). In keeping with an intense positive selection pressure for change, substitution rates obtained by comparing homologous exons are high, whereas introns sequences are remarkably conserved. In exons, non-synonymous substitutions exceed synonymous substitutions. |  |
| The present paper describes the application of FORS-D analysis to these sequences. Contrary to the assertion that for venom PLA2 genes "the regions corresponding to introns of precursor RNAs involved no significant secondary structure" (Nakashima et al. 1993), FORS-D analysis indicates an evolutionary pressure to conserve intron sequences in order to maintain the potential for secondary structure, and hence the potential to undergo recombination. | |
|
FORS-D analysis. This evaluates base order-determined stem-loop potential, which closely corresponds to the "statistically significant" stem-loop potential of Le and Maizel (1989). The theory and application of the method were described previously (Forsdyke, 1995a). Determination of substitution density. Sequences of different PLA2 genes were aligned with the program GENALIGN using the "regions" method, a matching weight of 1.0, and a deletion weight (gap penalty) of -0.5 (Martinez, 1988). The program was accessed through the Bionet on-line computing service (IntelliGenetics Inc., Mountain View, California). Base substitutions and "indels" (insertions or deletions) were counted in successive 200 nt windows, each of which overlapped the preceding window by 150 nt. Substitutions dominated the alignments and changes in deletion weight over a range from -0.25 to -1.0 caused few interchanges between substitutions and indels. Thus, the general patterns of substitution and indel densities were independent of deletion weight. No corrections were made for the possibility of multiple substitutions. High substitution density in exons. Many species have multiple PLA2 genes, which usually consist of four exons (Davidson and Dennis, 1990). In snakes a subset of these genes encodes proteins which contribute to the toxicity of venom. Intraspecies comparisons of base substitutions in snake venom phospholipase genes show high substitution densities in exons, and low substitution densities in introns. This applies to the habu snake (Nakashima et al, 1993), and to the rattlesnake (John et al. 1994). |
|
|
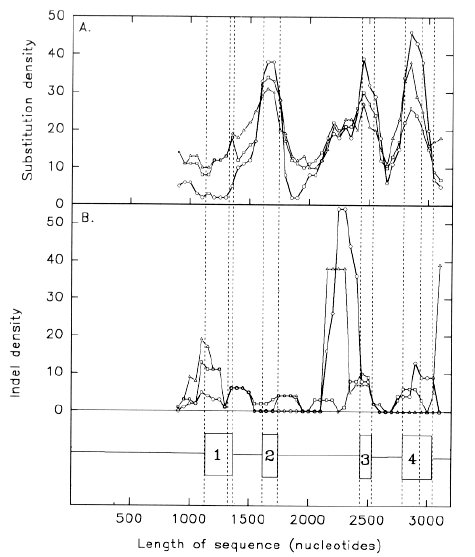
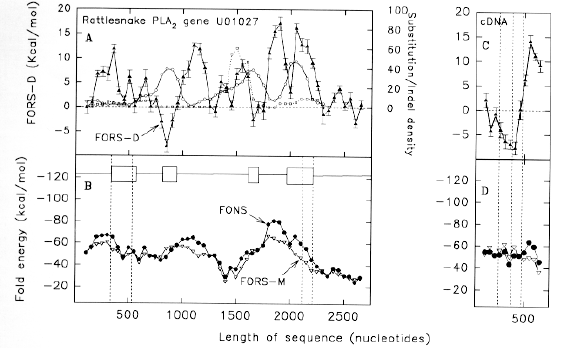
| FIG. 1.-Distribution of (A) substitutions and (B) indels in rattlesnake PLA2 acidic subunit gene U01026, relative to rattlesnake PLA2 basic subunit gene U01027 (circles), habu snake PLA2 gene TFLPA2P1 (squares), and habu snake PLA2 gene TFLPA2P2 (triangles). Substitutions and indels are counted in successive 200 nt windows, each of which overlaps the preceding window by 150 nt. Data points at 50 nt intervals each correspond to the centre of a window. The positions of the four exons are shown as boxes in B. Vertical dashed lines indicate exon borders, the beginning of the protein-coding part of exon 1, and the end of the protein-coding part of exon 4. |
|
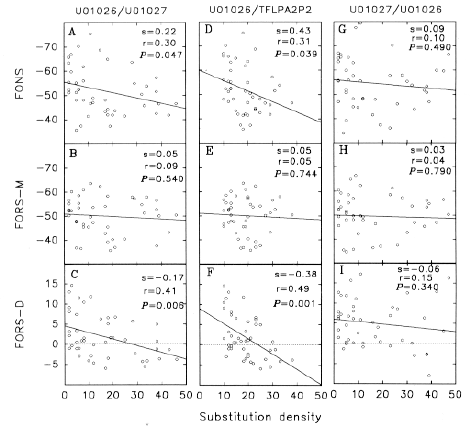
Figure 1a demonstrates this, both within species, and between species. Substitutions are scored for successive 200 nt windows which overlap by 150 nt. Relative to the rattlesnake PLA2 acidic subunit gene (GenBank designation U01026), the rattlesnake PLA2 basic subunit gene (GenBank designation U01027), and two habu snake PLA2 genes (GenBank designations TFLPA2P1 and TFLPA2P2), show many substitutions in all but the first exon, most of which does not code for protein. In the fourth exon, substitution density is greatest in the protein-encoding region. Because some of the 200 nt windows, centred in introns, overlap exons, sharp demarkations between the low substitution density introns and high substitution density exons, are not apparent. However, the relatively low intron substitution density is not uniform in the second intron; here the density increases in the 3' half of the intron. Essentially similar density patterns are obtained when different venom PLA2 genes are taken as a basis for comparison (e.g. TFLPA2P2 compared with U01026, U01027 and TFLPA2P1; data not shown). Thus, although the particular bases substituted tend to be characteristic of a particular pair of genes, the overall substitution patterns are unlikely to be random. Figure 1b shows similar data for base insertions and deletions ("indels"). Indels are in both coding and non-coding regions. Major peaks occur at the 3' end of the second intron in the cases of the U01026/U1027 and U1026/TFLPA2P2 comparisons. In the former case the peak is associated with a 38 nt CG-rich sequence in U01026, which is also present in the habu snake genes, but not in U01027. In the latter case the peak is associated with a microsatellite repeat (CA)n in TFLPA2P2, which is not so evident in the other snake venom PLA2 genes. The indel density patterns shown in figure 1b are essentially similar to those obtained when different venom PLA2 genes are taken as a basis for comparison (data not shown). Thus, although the actual bases inserted or deleted tend to be characteristic of a particular pair of genes, the overall patterns are unlikely to be random.
Reciprocal relationship between substitution density and FORS-D. The 200 nt windows used to characterize substitution and indel densities (fig. 1), were subjected to the program FOLD (Zuker, 1989) to determine the energetically most favourable folded structures. |
|
|
| FIG. 2.-Comparison of distribution of values for FONS (closed circles), FORS-M (open triangles) and FORS-D (closed triangles), with distribution of base substitutions (open circles) and indels (open squares), for the rattlesnake PLA2 acidic subunit gene U01026 (A, B). Values are for the same 200 nt windows as shown in figure 1. Substitutions and indels are relative to the rattlesnake PLA2 basic subunit gene U01027. Boxes in B indicate the location of the four exons, with four fine vertical lines showing the beginning of exon 1, the beginning of the protein-coding part of exon 1, the end of the protein-coding part of exon 4, and the end of exon 4. On the right (C,D) are shown FONS, FORS-M and FORS-D values for the cDNA. Vertical lines indicate splice sites (exon-intron junctions). |
|
|
| FIG. 3.-Comparison of distribution of values for FONS, FORS-M and FORS-D, with distribution of base substitutions and indels, for the rattlesnake PLA2 basic subunit gene U01027. Substitutions and indels are relative to the rattlesnake PLA2 acidic subunit gene U01026. Symbols and other details are as in figure 2. |
|
In figures 2b (rattlesnake gene U01026) and 3b (rattlesnake gene U01027), the minimum free energies associated with each structure (FONS values) are shown together with the mean values for ten randomized versions of the same sequence windows (FORS-M values). In keeping with previous observations on a variety of genes (Forsdyke, 1995a), FONS values tend to be more negative than FORS-M values (indicating greater stability of the secondary structure of the natural sequence). This is particularly apparent in the 5' non-coding part of exon 1, in the first part of intron 2, and in intron 3. The differences (FORS-M less FONS) are expressed in figures 2a and 3a as the FORS-D ("folding of randomized sequence difference") values. Positive FORS-D values reflect a contribution of base order (primary sequence), rather than of base composition, to the stability of the optimum structure. The previously noted tendency for protein-encoding parts of exons to have zero or negative FORS-D values, and for introns to have positive FORS-D values (Forsdyke, 1995a), is generally apparent. In the case of U01026 the first intron has low values, but this is not seen in the case of U01027. In the case of U01027 the coding part of the fourth exon corresponds to a relative decline in FORS-D value, but remains positive. It should be noted that high FORS-D values, reflecting local adaptation of base order for the generation of secondary structure, are not necessarily correlated with high FONS values (e.g. high FORS-D values occur in the 3' half of the 2nd intron and in the 3' non-coding part of the fourth exon). Figures 2c,d and 3c,d show FONS, FORS-M and FORS-D values for U01026 and U01027 (rattlesnake) cDNAs. Fine vertical lines indicate sites of intron removal. FORS-D values are low in the regions corresponding to the middle two exons, but increase in the 5' and 3' exons, which contain extensive non-coding regions. Figures 2a and 3a also show substitution and indel densities taken from figure 1. There is an approximately reciprocal relationship between FORS-D values and substitution density. Where FORS-D values are low (coding parts of exons), substitution densities are high. Where FORS-D values are high (introns; 5' and 3' non-coding regions), substitution densities are low. However, the 3' end of the 2nd intron, where indel density is highest, is characterized by relatively high values both for FORS-D and substitution density. Similar results were obtained when the other venom PLA2 genes under study were compared in this manner. More objective evidence for a relationship between secondary structure and substitution density was provided by plotting individual window values for FONS, FORS-M and FORS-D, against the corresponding substitution densities. For each pair of genes three linear regression plots were obtained. |
|
|
| FIG. 4.-Linear regression analysis of the relationship between FONS, FORS-M and FORS-D values (kcal/mol), and substitution density (base exchanges/200 nt window) in snake venom PLA2 genes. For the left column of figures (A,B,C), data are from figure 2 (FONS, FORS-M and FORS-D values for rattlesnake PLA2 acidic subunit gene U01026, and substitutions relative to rattlesnake PLA2 basic subunit gene U01027). The middle column (D,E,F) shows FONS, FORS-M and FORS-D values for rattlesnake PLA2 acid subunit gene U01026, and substitutions relative to habu snake PLA2 gene TFLPA2P2. For the right column (G,H,I) data are from figure 3 (FONS, FORS-M and FORS-D values for rattlesnake PLA2 basic subunit gene U01027, and substitutions relative to rattlesnake PLA2 acidic subunit gene U01026). Parameters of the least-squares line shown in each figure are, slope (s), the correlation coefficient (r), and the probability that the slope of the line is not significantly different from zero (P). |
| Figure 4 shows results for three gene pairs (U01026/U01027, U01027/U01026, U01026/TFLPA2P2). In general, plots for FORS-M are horizontal, plots for FONS slope down slightly and plots for FORS-D slope down more steeply. Within each figure are listed values for the slope (s), correlation coefficient (r), and the probability (P) that the slope is not significantly different from zero. The latter two values are most significant (e.g. high values for r and low values for P) in the case of plots of FORS-D against substitution density, and least significant in the case of plots of FORS-M against substitution density. The results of similar plots for other gene combinations are shown in Table 1. Although values for some gene combinations have relatively low r values and high P values (e.g. figs. 4g,h,i), direct inspection of the corresponding plots of values as a function of sequence position (e.g. fig. 3a) tends to support the generalization that, in most parts of a gene, there is a reciprocal relationship between substitution density and FORS-D value. |
| Snake GenBank Name |
RATTLESNAKE | HABU SNAKE | |||||||||||
| U01026 | U01027 | TFLPA2P1 | TFLPA2P2 | ||||||||||
| . | . | sl | r | P | sl | r | P | sl | r | P | sl | r | P |
| RATTLESNAKE | . | ||||||||||||
| U01026 . |
FONS | . | 0.22 | 0.30 | 0.047 | 0.32 | 0.24 | 0.112 | 0.43 | 0.31 | 0.039 | ||
| FORS-M | 0.05 | 0.09 | 0.540 | 0.01 | 0.01 | 0.946 | 0.05 | 0.05 | 0.744 | ||||
| FORS-D | -0.17 | 0.41 | 0.006 | -0.31 | 0.42 | 0.004 | -0.38 | 0.49 | 0.001 | ||||
| U01027 . |
FONS | 0.09 | 0.10 | 0.490 | . | 0.13 | 0.17 | 0.244 | 0.02 | 0.02 | 0.920 | ||
| FORS-M | 0.03 | 0.04 | 0.790 | 0.11 | 0.18 | 0.222 | -0.09 | 0.10 | 0.504 | ||||
| FORS-D | -0.06 | 0.15 | 0.340 | -0.02 | 0.06 | 0.692 | -0.10 | 0.19 | 0.216 | ||||
| HABU SNAKE | . | ||||||||||||
| TFLPA2P1 . |
FONS | 0.28 | 0.22 | 0.140 | 0.02 | 0.06 | 0.693 | . | 0.26 | 0.23 | 0.132 | ||
| FORS-M | -0.03 | 0.03 | 0.817 | 0.01 | 0.02 | 0.980 | 0.04 | 0.05 | 0.757 | ||||
| FORS-D | -0.32 | 0.47 | 0.001 | -0.02 | 0.09 | 0.550 | -0.22 | 0.35 | 0.017 | ||||
| TFLPA2P2 . |
FONS | 0.10 | 0.06 | 0.703 | 0.01 | 0.01 | 0.940 | 0.21 | 0.15 | 0.336 | . | ||
| FORS-M | 0.02 | 0.01 | 0.932 | -0.02 | 0.02 | 0.889 | 0.08 | 0.08 | 0.618 | ||||
| FORS-D | -0.08 | 0.11 | 0.478 | -0.03 | 0.06 | 0.705 | -0.13 | 0.20 | 0.182 | ||||
| NOTE. - Linear regression analyses, examples of which are shown in fig.4, were performed for various pairs of snake venom PLA2 genes. FONS, FORS-M, and FORS-D values were obtained for the vertically listed genes. Substitution densities were relative to the horizontally-listed genes. For a particular gene pair, and either the FONS, FORS-M, or FORS-D value, the set of three numbers (sl/r/P) refers to the slope of the linear regression plot (sl), the correlation coefficient (r), and the probability that the slope is not significantly different from zero (P). | |||||||||||||
|
Although unusual, high conservation of non-coding sequences in cDNAs, to an extent exceeding that of neighbouring coding sequences, has been observed both within species (Blum et al. 1990), and between species (Duret et al. 1993). Ogawa and coworkers (1992) compared PLA2 cDNAs of the habu snake (Trimeresurus flavoviridis), which were found to have extremely high conservation of 5' and 3' non-coding sequences (98% and 89% identities, respectively). They correlated this with high stem-loop potential, and suggested an important functional role at the translation level. They also noted high conservation of the nucleotide sequence corresponding to the 16 amino acid signal peptide. However, the remaining protein-encoding sequence showed low conservation (67% identity), mainly due to nucleotide substitutions. An unusually high proportion of these were non-synonymous, thus changing the protein sequence. These observations were consistent with strong selection pressures both to conserve the signal peptide function, and to change the function of the mature protein (122 amino acids). The change would be required to counter adaptations to resistance of the prey. The signal peptide would, of course, be discarded and would not interact with the prey. PLA2 generates a cytotoxic product (Takeda et al. 1982), and reacts with broad specificity binding proteins (Lambeau et al. 1994), which might adapt to generate resistance. A similar evolutionary process (positive selection) appears to operate in the cases of:
When habu snake PLA2 genomic sequences became available, Nakashima and coworkers (1993) found that intron sequences were as highly conserved as the 5' and 3' non-coding regions. This made explanations of functional effects at the translation level unlikely, and raised the possibility of effects of nucleic acid secondary structure at the genomic level, or at the level of RNA processing. However, Nakashima and coworkers found no secondary structure when the sequences were analyzed by the method which had been used to show secondary structure in 5' and 3' non-coding regions (Zuker and Steigler, 1981; Ogawa et al. 1992). The observations on habu snake venom PLA2 genes were also found to apply to the venom PLA2 genes of the Mojave rattlesnake (Crotalus scutulatus scutulatus). John and coworkers (1994) pointed out that "the greater intron identity appears to violate evolutionary dogma, which predicts greater divergence in the non-coding regions". They suggested a critical role of introns in pre-mRNA stabilization or processing. The present work began by demonstrating similar patterns of conservation, whether comparisons were made within species or between species (fig. 1). The patterns were similar for different gene pairs, but the actual changes tended to be characteristic of a particular gene pair. Thus, although gene duplication generating multiple venom PLA2 genes may have occurred in an ancestor prior to the divergence of the habu snake and rattlesnake lineages (Davidson and Dennis, 1990), most sequence differentiation is likely to have occurred within species, after the divergence. Similarity of patterns of different species indicates similar selective constraints. Direct observations of the folding into stem-loop structures of sections of the sequences (FONS values) indicated some increased secondary structure potential in introns, relative to exons (figs. 2b, 3b). This was particularly apparent in the third intron. The failure of others to note this may reflect the use in the present work of an improved folding algorithm (Zuker, 1989), and better energy values for base stacking and loop destabilization (Turner et al. 1988). However, FONS values reflect the contribution of both base order and base composition. Base composition probably serves genome-wide or genome-sector specific functions, rather than gene-specific functions, as discussed elsewhere (Forsdyke, 1995a-c). Evolutionary effects on the base order became apparent when the negative FONS values (greater negativity reflecting more stable structures) were subtracted from the negative FORS-M values (which reflect the contribution of base-composition alone). This produced FORS-D values. Positive FORS-D values, indicating the acceptance of mutations which contribute to the stability of secondary structure by changing base order, were generally present both in introns and in 5' and 3' non-coding regions. Thus, the structures in these regions are likely to be functionally relevant, although they may not necessarily be correlated with high FONS values. Zero and negative FORS-D values, reflecting no acceptance of such mutations, were generally present in the exons encoding the mature PLA2 protein (figs. 2a, 3a). Thus, the conservation of intron sequences could reflect a functional constraint for which base order-determined stem-loop potential is required. The direct relationship between base order-determined stem-loop potential and sequence conservation (i.e. inverse relationship to substitution density), was shown by linear regression analysis (fig. 4; table 1). Similar observations have been made on retroviral (HIV-1) sequences (Le et al. 1988, 1989; Forsdyke, 1995d).
Indels probably play an important role in interfering with the initial alignment (register) of stem-loops in potentially recombining homologous strands (Tomizawa, 1993). Although, within a genome, the introns for different venom PLA2 genes may show great conservation (tending to provoke recombination), indels would make it more likely that recombination would occur only between identical genes. The differences in exon sequences would also ensure that, even if the initial "kissing" interaction between the loops of stem-loops were stable (Kleckner and Weiner, 1993; Tomizawa, 1993), subsequent consummation of the union would be unlikely (Rayssiguier et al. 1989; Radman and Wagner, 1993).
The results presented here also suggest that it would be of considerable interest to extend the analysis to other genes under positive selection pressure (Hill and Hastie, 1987; Hughes and Nei, 1989), as has been done in the case of retroviral genomes (Forsdyke, 1995d). Indeed, preliminary studies show that regions encoding peptide-binding sites in MHC genes have negative FORS-D values [Forsdyke 1996b unpublished in 1995]. It has been noted that troponin-C genes are under positive Darwinian selection (Ohta, 1994), and that certain exons show very marked negative FORS-D values (Forsdyke, 1995a,c[=1996a]). It would also be of interest to compare venom PLA2 genes, with other PLA2 genes which would not be under such pressure. Acknowledgements. This work was supported by a grant from the Medical Research Council of Canada. |
|
LITERATURE CITED BERNSTEIN, C., and H. BERNSTEIN. 1991. Aging, Sex and DNA Repair. Academic Press, San Diego. BLUM, S., R. E. FORSDYKE, and D. R. FORSDYKE. 1990. Three human homologs of a murine gene encoding an inhibitor of stem cell proliferation. DNA Cell Biol. 9:589-602. BRANNAN, C. I., E. C. DEES, D. S. INGRAM, and S. M. TILGHMAN. 1990. The product of the H19 gene may function as an RNA. Mol. Cell. Biol. 10:28-36. BROCKDORFF, N., A. ASHWORTH, G. F. KAY, V. M. McCABE, D. P. NORRIS, P. J. COOPER, S. SWIFT, and S. RASTAN. 1992. The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71:515-526. DAVIDSON, F. F., and E. A. DENNIS. 1990. Evolutionary relationships and implications for the regulation of phospholipase A2 from snake venom to human secreted forms. J. Mol. Evol. 31:228-238. DURET, L., F. DORKELD, and C. GAUTIER. 1993. Strong conservation of non- coding sequences during vertebrate evolution: potential involvement in post- transcriptional regulation of gene expression. Nucleic Acids Res. 21:2315-2322. FORSDYKE, D. R. 1991. Early evolution of MHC polymorphism. J. Theor. Biol. 150:451-456. FORSDYKE, D. R. 1994. Relationship of X chromosome dosage compensation to intracellular self/not-self discrimination: a resolution of Muller's paradox? J. Theor. Biol. 167:7-12. FORSDYKE, D. R. 1995a. A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol. Biol. Evol. 12:949-958. FORSDYKE, D. R. 1995b. Relative roles of primary sequence and (G + C)% in determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J. Mol. Evol. 41:573-581. FORSDYKE, D. R. 1995c. Different biological species "broadcast" their DNAs at different (G+C)% "wavelengths". (submitted) -- J. Theor. Biol. (1996a) 178:405-417. FORSDYKE, D. R. 1995d. Reciprocal relationship between stem-loop potential and substitution density in retroviral quasispecies under positive Darwinian selection.(submitted) -- J. Mol. Evol.(1995) 41:1022-1037. HARDING, K. A., and K. R. G. WELCH. 1980. Venomous Snakes of the World. Pergamon Press, Oxford. HAWKINS, J. D. 1988. A survey of intron and exon lengths. Nucleic Acids Res. 16:9893-9905. HILL, R. E., and N. D. HASTIE. 1987. Accelerated evolution in the reactive centre regions of serine protease inhibitors. Nature 326:96-99. HUGHES, A. L., and M. NEI. 1989. Nucleotide substitution at major histocompatability complex class II loci: evidence for overdominant selection. Proc. Natl. Acad. Sci. USA. 86:958-962. JOHN, T. R., L. A. SMITH, and I. I. KAISER. 1994. Genomic sequences encoding the acidic and basic subunits of Mojave toxin: unusually high sequence identity of non-coding regions. Gene 139:229-234. JOYCE, G. F., and L. E. ORGEL. 1993. Prospects for understanding the origin of the RNA world. Pp 1-25 in R. F. GESTELAND, and J. F. ATKINS, eds. The RNA World. Cold Spring Harbour Laboratory Press, New York. KLECKNER, N., R. PADMORE, and D. K. BISHOP. 1991. Meiotic chromosome metabolism: one view. Cold Spring Harbour Symp. Quant. Biol. 56:729-743. KLECKNER, N. and B. M. WEINER. 1993. Potential advantages of unstable interactions for pairing of chromosomes in meiotic, somatic and premeiotic cells. Cold Spring Harbour Symp. Quant. Biol. 58:553-565. LAMBEAU. G., P. ANCIAN, J. BARHANIN, and M. LAZDUNSKI. 1994. Cloning and expression of a membrane receptor for secretory phospholipase A2. J. Biol. Chem. 269:1575-1578. LE, S-Y., J-H. CHEN, M. BRAUN, M. GONDA, and J. V. MAIZEL.1988. Stability of RNA stem-loop structure and distribution of non-random structure in the human immunodeficiency virus (HIV-1). Nucleic Acids Res. 16:5153-5168. LE, S-Y., J-H. CHEN, D. CHATTERJEE, and J. V. MAIZEL. 1989. Sequence divergence and open regions of RNA secondary structures in the envelope regions of the 17 human immunodeficiency virus isolates. Nucleic Acids Res. 17:3275-3288. LE, S-Y. and J. V. MAIZEL. 1989. A method for assessing the statistical significance of RNA folding. J. Theor. Biol. 138:495-510. MARTINEZ, H. M. 1988. A flexible multiple sequence alignment program. Nucleic Acids Res. 16:1683-1691. NAKASHIMA, K-I., T. OGAWA, N. ODA, M. HATTORI, Y. SAKAKI, H. KIHARA, and M. OHNO. 1993. Accelerated evolution of Trimeresurus flavoviridis venom gland phospholipase A2 isozymes. Proc. Natl. Acad. Sci. USA 90:5964-5968. OGAWA, T., N. ODA, K-I NAKASHIMA, H. SASAKI, M. HATTORI, Y. SAKAKI, H. KIHARA, and M. OHNO. 1992. Unusually high conservation of untranslated sequences in cDNAs for Trimeresurus flavoviridis phospholipase A2 isoenzymes. Proc. Natl. Acad. Sci. USA 89:8557-8561. OHTA, T. 1994. Further examples of evolution by gene duplication revealed through DNA sequence comparisons. Genetics 138:1331-1337. RADMAN, M., and R. WAGNER. 1993. Mismatch recognition in chromosomal interactions and speciation. Chromosoma 102:369-373. RAYSSIGUIER, C., D. THALER, and M. RADMAN. 1989. The barrier to recombination between Echerichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342:396-401. ROMANOVA, L. I., V. M. BLINOV, E. A. TOLSKAYA, E. G. VIKTOROVA, M. S. KOLESNIKOVA, E. A. GUSEVA, and V. I. AGOL. 1986. The primary structure of crossover regions of intertypic poliovirus recombinants: a model of recombination between RNA genomes. Virology 155:202-213. SCPAER, E. G., and J. I. MULLINS. 1993. Rates of amino acid change in the envelope protein correlates with pathogenicity of primate lentiviruses. J Mol Evol 37:57-65 STOLTZFUS, A., D. F. SPENCER, M. ZUKER, J. M. LOGSDON, and W. F. DOOLOTTLE. 1994. Testing the exon theory of genes: the evidence from protein structure. Science 265:202-207. TAKEDA, A., R. G. E. PALFREE, and D. R. FORSDYKE. 1982. Role of serum in inhibition of cultured lymphocytes by lysophosphatidylcholine. Biochem. Biophys. Acta 710:87-98. TOMIZAWA, J. 1993. Evolution of functional structures of RNA. Pp. 419-445 in R. F. GESTELAND, and J. F. ATKINS, eds. The RNA World.Cold Spring Harbour Laboratory Press, New York. TURNER, D. H., N. SUGIMOTO, and S. M. FREIER. 1988. RNA structure prediction. Annu. Rev. Biophys. Chem. 17:167-192. ZUKER, M. 1989. Computer prediction of RNA secondary structure. Meth. Enzym. 180:262-289. ZUKER, M., and P. STEIGLER. 1981. Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucleic Acids Res. 9:133-148. |
Single Sequence Method
Stem-loop
potential in MHC genes:
a new way of evaluating positive Darwinian selection?
D. R. Forsdyke
(1996b) Immunogenetics 43:
182-189.
[With copyright permission from Springer Inc.]
Abstract
Introduction
Methods
Results
DiscussionDecreased FORS-D values as an indicator of positive selection?
Intron conservation as an index of positive selection?
Evolutionary aspects
| Abstract The domains of polymorphic MHC proteins which interact with peptides and T cell receptors are considered to have been under positive evolutionary selection pressure. Evidence for this is a high ratio of non-synonymous to synonymous mutations in the corresponding genomic domains. By this criterion snake venom phospholipase A2 genes have also been under positive selection pressure. Recent studies of the latter genes indicate that positive selection has overridden an evolutionary pressure on base order which normally promotes the potential to extrude single strand stem-loops from supercoiled duplex DNA ("fold pressure"). This has resulted in base order-dependent stem-loop potential being shifted to introns, which are highly conserved between species. It is now shown that, like snake venom phospholipase A2 genes, the domains of polymorphic MHC genes which appear to have responded to positive selection pressure have decreased base order-dependent stem-loop potential. The evolutionary pressure to generate stem-loop potential (believed to be important for recombination) has been overridden less in exons under negative "purifying" selection, than in exons under positive "Darwinian" selection. Thus, base order-dependent stem-loop potential shows promise as an independent indicator of positive selection. |
![]()
|
Class I and class II major histocompatibility complex (MHC) proteins bind intracellular peptides derived from intracellular and imported extracellular proteins and present them at the cell surface to T cell receptors (Bjorkman and Parham, 1990). Intracellular and extracellular pathogens which mutate their proteins to evade this host immune defence, present an evolutionary "moving target" to which the host species must respond by corresponding mutations in the peptide-recognition domains of MHC proteins (Klein and O'hUigin, 1994). In these regions advantageous mutations are positively selected ("Darwinian selection"), whereas in other regions the sequence is conserved, most mutations being disadvantageous and hence being negatively selected ("purifying selection"). Evaluating which gene and protein domains of a pathogen and its host are under positive or negative evolutionary selection is an important step leading to the identification of possible molecular sites for therapeutic attack. A widely accepted method of determining if a region of a protein has been under positive selection pressure is to calculate for the corresponding nucleic acid the ratio of non-synonymous mutations (which change the amino acid sequence) to synonymous mutations (which do not change the amino acid sequence). A high proportion of non-synonymous mutations indicates that a region has been under positive selection. This has been shown to apply to the peptide-binding regions of both class I and class II MHC proteins (Hughes and Nei, 1988,1989). The method has also been used to provide evidence for positive selection in the cases of the active sites of protease inhibitors believed to be under the selection pressure of pathogen-derived proteases (Hill and Hastie, 1987), the variable regions of immunoglobulins under selection pressure of extracellular pathogens (Tanaka and Nei, 1989), the surface-exposed loops of pathogen proteins under the selection pressure of host immune defences (Smith et al. 1995), and snake venom phospholipase A2 molecules under the selection pressure of resistant prey/predators. In the latter case the entire secreted protein appears to have been the subject of positive selection (Nakashima et al. 1993). In such studies, "since synonymous changes do not change the amino acid, synonymous mutations are expected to be neutral, or nearly so" (Hughes and Hughes, 1995). However, there is growing evidence that many "neutral" mutations reflect the outcome of two major evolutionary pressures, "GC/AT pressure", and "fold pressure"; these together generate stem-loop potential (Forsdyke, 1995a-c; 1996a,b). The former is assessed as the base composition-determined component ("FORS-M" value), and the latter as the base order-determined component ("FORS-D" value), of the total stem-loop potential of a nucleic acid segment ("FONS" value). The sequence of an exon appears to reflect an evolutionary compromise between these two pressures and the pressure to encode an optimum amino acid sequence ("protein pressure"). In the case of genes under intense positive selection, protein pressure would be expected to dominate, with adverse consequences for the other pressures. This would apply particularly to fold pressure which, in contrast to GC/AT pressure, is a local evolutionary "strategy", rather than a genome or genome-sector strategy (Forsdyke, 1995a).
Recent studies of the distribution of stem-loop potential in snake venom phospholipase A2 genes have shown that fold pressure has been strongly subverted in regions under positive Darwinian selection, suggesting a new approach to the evaluation of positive selection (Forsdyke, 1995b). This paper explores the applicability of the approach to MHC genes. |
|
The theoretical basis of the evaluation of stem-loop potentials in nucleic acid sequences ("FORS-D analysis") is described elsewhere (Le and Maizel, 1989; Forsdyke, 1995a-c). The Genetics Computer Group implementation of the program RNAFOLD, was accessed as part of the Molecular Biology Data Service of the National Research Council, Ottawa (Zuker, 1989). The program was designed for the study of secondary structure in non-supercoiled single-stranded RNA molecules. It is assumed here that it is applicable to DNA. This assumption is not entirely valid (Breslauer et al., 1986). For example, the 2' hydroxyl group in RNA can contribute of the order of 1 kcal/mol to helix stability. Since the present study was mainly concerned with differences in fold energies, rather than absolute values, differences between RNA and DNA are considered of minor importance. Following the completion of the study, Nielsen and coworkers (1995) made available local data files for folding DNA for use with the program LRNA, which is an advanced version of RNAFOLD (Jaeger et al., 1990). Comparison of the outputs of different programs confirms the assumption that the distribution of fold values along genes shows the same general pattern with different programs when using local data files either for RNA or for DNA (Forsdyke, unpublished work). RNAFOLD is applied to natural and randomized versions of consecutive overlapping "windows" in nucleic acid sequences to generate, respectively, "folding of natural sequence" (FONS) values, and "folding of randomized sequence" (FORS) values. The FONS value for a window is subtracted from the corresponding mean of several FORS values (FORS-M), to generate the "folding of randomized sequence difference" (FORS-D) value for the window. The theory of stem-loop potentials (Forsdyke, 1995a-c) predicts that, in local regions of the genome, FORS-M values (the base composition-dependent component of the FONS value) should be constant. However, cyclical fluctuations with a period around 0.6 kb are seen in most genes. While this may reflect some fundamental aspect of chromatin organization, it is possible that low negative FORS-M values, usually corresponding to AT-rich regions, are the first to extrude from negatively supercoiled duplex DNA (Sinden, 1994). These AT-rich regions might be close to the centres of "minimum efficient processing segments" involved in recombination (Radman et al. 1993). The theory also implies that, whereas zero FORS-D values indicate failure to accommodate to evolutionary pressure for stem formation, negative FORS-D values indicate that a sequence has been modified to oppose stem formation (i.e. promote duplex stability; Forsdyke, unpublished work [see Forsdyke 1998]). This might work at the DNA level to prevent recombination. Alternatively, it might work at the mRNA level to prevent translational delay. |
| Figure 1 shows an example of FORS-D analysis as applied to an allele of a highly polymorphic human MHC class I gene, the products of which would be expressed on the surface of most nucleated somatic cells. |
|
|
Fig. 1. FORS-D analysis of an allele of a highly polymorphic human MHC class I gene (HLA-A1; GenBank name HSHLAA1). Optimum secondary structures of minimum free energy were calculated for successive 200 nt windows, each overlapping the previous window by 175 nt. Points correspond to the middle of each window. The distribution of FORS-D values (± standard error) are shown in the upper part of the figure. Positive values indicate a positive contribution to the stability of stem-loop structures. The lower part of the figure shows the distribution of the FONS and FORS-M values, from which the FORS-D values were derived by subtraction. High negative values (kcal/mol) of FONS and FORS-M correspond to high stability. By subtracting FONS values from FORS-M values, it is arranged that FORS-D values will usually be positive. Exons positions are shown as numbered boxes. Exon 1 encodes the signal peptide. Exons 2-4 encode the three extracellular protein domains (a1, a2, a3). Exon 5 encodes the transmembrane domain. Exons 6 and 7 encode intracellular domains. Dark areas in the boxes of exons 2 and 3 correspond to binding-cleft amino acid residues which contact peptide or T cell receptor. |
|
The lower part of the figure shows the distribution of the FONS and FORS-M values from which the FORS-D values were calculated by subtraction (FORS-M - FONS). As found in most other genes studied (Forsdyke, 1995a), positive FORS-D values predominate, especially in introns and flanking regions. Extremely negative FORS-D values are associated with the first four exons (shown as numbered boxes). The first exon encodes the signal peptide. The second and third exons encode the highly polymorphic a1 and a2 domains of the protein. When inter- and intra-species comparisons are made with other class I genes, these two exons show unusually high proportions of non-synonymous base substitution mutations in regions corresponding to the variable amino acid residues which interact with peptides and T cell receptors (shown as black boxes within the exon boxes; Hughes and Nei, 1988; Bjorkman and Parham, 1990). The fourth exon encodes the non-polymorphic a3 domain, which associates non-covalently with b2-microglobulin. The negative FORS-D region here is not so broadly spread out as in the second and third exons, and includes the sequence encoding the conserved CD8 recognition domain (Salter et al. 1990). Decreased FORS-D values in exons indicate insufficient evolutionary flexibility in usage of synonymous codons and in the exchange of amino acids with similar functions to permit base order to optimize simultaneously both coding potential and stem-loop potential (Forsdyke, 1995a). Decreased FORS-D values are likely to have resulted from positive selection in the case of the second and third exons, and negative ("purifying") selection in the case of the conserved fourth exon. |
|
|
| Fig. 2. FORS-D analysis of the second exon of the human MHC class I gene shown in Figure 1, using successive 200 nt windows overlapping by 195 nt. Green boxes indicate correspondence with amino acid residues which contact peptide, or the T cell receptor (asterisks; Bjorkman and Parham, 1990). Vertical dashed lines indicate exon-intron boundaries. |
| The values shown in Figure 1 are for consecutive 200 nt windows each of which overlapped the preceding window by 175 nucleotides. Thus a window is displaced by 25 nt from the preceding window. Figure 2 shows data for the 2nd exon alone using windows overlapping by 195 nt. Thus a window is displaced by only 5 nt from the preceding window. The results at this higher level of resolution confirm the tendency for low FORS-D values to associate with regions involved in the binding of peptide and/or T cell receptor (i.e. regions under positive selection pressure). |
|
|
| Fig. 3. FORS-D analysis of an allele of a highly polymorphic mouse MHC class II b chain gene (H2-Ab; GenBank MUSMHABZ2). Exons 2 and 3 correspond to extracellular protein domains. Exon 2 is the polymorphic domain which interacts with peptide and the T cell receptor. Exon 4 corresponds to the transmembrane protein domain. Exon 5 corresponds to an intracellular protein domain. Other details are as in Figure 1. |
|
|
| Fig. 4. FORS-D analysis of exon 2 of the mouse MHC class II b chain gene shown in Figure 3. Black boxes indicate regions encoding amino acids which interact with peptide and/or the T cell receptor. The striped box indicates the region encoding amino acids involved in dimer interaction. |
| Figures 3 and 4 show data similar to those of Figures 1 and 2, for part of a polymorphic mouse MHC class II Ab chain-encoding gene (Hughes and Nei, 1989). In this case only the second exon encodes the variable sequences which interact with peptide and T cell receptor, and this exon shows much lower FORS-D values than the third exon. The association of decreased FORS-D values with the second exon is evident both at "low" resolution (Fig. 3; windows moving in steps of 25 nt), and at "high" resolution (Fig. 4; windows moving in steps of 5 nt). Again, there is a tendency for decreased FORS-D values to associate with regions involved in the binding of peptide and/or T cell receptor. |
| Species | MHC class |
MHC gene |
Degree of poly- morph- ism |
Peptide binding | Genbank Name |
Reference | |||
| . | + Exon 2 |
+ Exon 3 |
- Exon 4 |
. | |||||
| Human | I | HLA-A1 | + + | -3.04+1.28(12) | -1.29+1.33(12) | -0.47+1.64(11) | HSHLAA1 | Girdle- stone 1900 |
|
| . | . | HLA-E | + | -1.81+1.45(12) | -2.39+1.32(12) | 0.25+1.04(12) | HUMMHEA | Koller et al. 1988 |
|
| . | . | HLA-H | - | 2.49+1.25(11) | -1.18+1.31(12) | -0.24+1.38(11) | HUM- MHANTLE |
Chorney et al. 1990 |
|
| . | . | HLA-J | - | -3.54+1.38(12) | -0.62+0.61(12) | 2.49+1.53(12) | HUM- MHHLAJC |
Messer et al. 1992 |
|
| . |
Peptide binding | . |
|||||||
| + Exon 2 |
- Exon 3 |
||||||||
| Human | II | HLA- DRA |
- | 1.20+1.30 (6) | 3.00+0.94 (6) | HSHL07 | Scham- boeck et al. 1983 |
||
| . | . | HLA- DRB4 |
+ + | -2.25+1.13 (6) | 2.21+1.13 (6) | HUM- MHDRW01 |
Anders- son et al. 1987 |
||
| . | |||||||||
| Mouse | II | H-2Ab | + + | -6.23+0.68 (12) | 6.64+0.78 (13) | MUS- MHABZ2 |
Mallissen et al. 1983 |
||
| . | . | H-2Ab2 | - | 0.96+0.99 (12) | 2.99+1.46 (12) | MUS- MHCAB1 |
Larhammer et al. 1985 |
||
| . | . | H-2Ea | - | -0.08+1.20 (11) | 2.63+1.36 (12) | MUS- MHIEAD |
Hyldig- Nielsen et al. 1983 |
||
|
Table 1. Average FORS-D values of exons encoding peptide-binding and non-peptide binding domains of MHC proteins. FORS-D values (kcal/mol) were determined for 200 nt windows, each of which overlapped the preceding window either by 175 nt (human class I and mouse class II), or by 150 nt (human class II). The number of windows is shown in parenthesis. The first and last windows corresponding to a particular exon were those whose centres were nearest to the 5' and 3' exon borders, respectively. Data for HLA-A1 and H2-Ab are from Figures 1 and 3 respectively. The average of the 13 differences between each peptide-binding exon and the corresponding non-peptide binding exon (-3.02 kcal/mol) is significantly different from zero (P<0.01). Comparing exons of the polymorphic genes (HLA-A1, HLA-E, HLA-DRB4, H2-Ab), with the corresponding exons of monomorphic controls (HLA-H for class I, HLA-DRA for HLADRB4, H2-Ab2 for H2-Ab), the average of the six differences (-3.63 kcal/mol) is significantly different from zero (P<0.02). HLA-J is not included in the latter calculation for reasons given in the text.
Table 1 summarizes similar studies with a variety of MHC genes. Average FORS-D values for windows in peptide binding exons are compared with average values for windows in non-peptide-binding exons (the fourth exon in the case of class I genes and the third exon in the case of class II genes). With the exception of the 2nd exon of the pseudogene HLA-H (see later), each peptide-binding exon has a lower average FORS-D value than the corresponding non-peptide-binding exon. Polymorphic exons are considered to have been under more intense positive selection than monomorphic exons, some of which include pseudogenes (Hughes, 1995). Polymorphic exons usually have lower average FORS-D values than monomorphic exons, especially in the case of class II genes. It should be noted that, although sometimes considered monomorphic, the class I gene HLA-E does show some polymorphism (Ohya et al. 1990). Pseudogenes are presumed to have escaped the selection pressure of pathogens (Hughes, 1995), so that potential peptide-binding exons would be expected to have higher average FORS-D values than peptide-binding exons of genes still under that pressure. This is found in the case of the class I gene HLA-H (also known as HLA-AR and HLA-54). Exon 2 has an average FORS-D value of 2.49 kcal/mol, contrasting dramatically with the corresponding value for the highly polymorphic HLA-A1 gene (-3.04 kcal/mol; Table 1). However, another pseudogene (HLA-J, HLA-59) has an average FORS-D value in exon 2 of -3.54. This is largely due to extreme negative values in the domains encoding the b-sheet contact residues of the peptide-binding cleft; the domains encoding contact residues associated with a-helices had higher FORS-D values (data not shown). Exon 3 of HLA-J had a relatively high average FORS-D value (-0.62 kcal/mol), consistent with designation as a monomorphic pseudogene. |
|
Decreased FORS-D values as an indicator of positive selection? The average FORS-D value for 200 nt windows in long human genomic segments containing much intergenic DNA is about 4 kcal/mol (Forsdyke, 1995a). Similar values are found in various other species, including those with compact genomes and little intergenic DNA (Forsdyke, 1995c). Thus, positive base order-dependent stem-loop potential, assessed as the FORS-D value, is a widespread and fundamental characteristic of genomes. It is proposed that protein-encoding regions have been under evolutionary pressure to accommodate to the potential by:
The existence of exons with zero and negative base-order dependent stem-loop potentials indicates impairment of accommodation. Among the series of genes initially studied, a human troponin-c gene was found to have the most significant association of negative FORS-D values with exons (Forsdyke, 1995a; 1996a). Unlike the other genes in that study, the troponic C gene is believed is believe to have been under positive Darwinian selection pressure following gene duplication (Ohta, 1994). Based on the proportions of non-synonymous and synonymous mutations, snake venom phospholipase A2 genes are also believed to have been under positive selection pressure (Nakashima et al., 1993), and also have a significant association of negative FORS-D values with exons (Forsdyke, 1995b). In these two cases entire exons seem to have been involved in positive selection. Is the association of zero or negative FORS-D values with regions under positive selection a general phenomenon? Furthermore, is FORS-D analysis sufficiently sensitive in the case of exons only parts of which have been under positive selection pressure? The present work shows that when positive selection has affected local sectors of exons, these sectors tend to have low FORS-D values (Figs. 1-4). This suggests that the relationship between decreased base order-dependent stem-loop potential and positive selection is a general one, and that the method is sufficiently sensitive. Studies with retroviral genes, which may also have been under positive selection pressure (Scpaer and Mullins, 1993), further support the generalization (Forsdyke, 1995d). Regions under negative selection pressure may show decreased base-order stem-loop potential (Forsdyke, 1995a). However, the decrease seems more profound, and more sustained, in sequences which have been under positive, rather than under negative, selection pressure. This was suggested by comparing the troponin C gene with other genes (Forsdyke, 1995a; 1996a), and is shown here by comparing, within a gene, exons encoding extracellular protein domains under positive selection and exons encoding extracellular protein domains under negative selection (the fourth exon in the case of class I MHC genes, and the third exon in the case of class II MHC genes; Table 1). In general, the results with monomorphic exons, some of which are in pseudogenes (Table 1), are also consistent with decreased selection pressure relative to polymorphic exons (i.e. higher average FORS-D values in monomorphic exons, which have been under less selection pressure; Hughes, 1995). How accurately the present method can distinguish between regions showing high and low proportions of non-synonymous mutations is not known. Preliminary studies with decreased window size indicate that this approach is not likely to increase accuracy. The FORS-D values plotted in Figures 1-4 correspond to the middle of 200 nt windows. Remarkably, although an entire window is involved in the calculation, and the folding may not be symmetrical, the middle of the window seems to correlate best with regions under positive selection. Intron conservation as an index of positive selection? It is proposed that positive stem-loop potentials have evolved under a selection pressure to improve recombination (Forsdyke, 1995a). The diversification of exons under positive selection pressure, would shift to introns the burden of developing stem-loop potential, so that they would then become highly conserved. Thus, introns are conserved in snake venom phospholipase A2 genes (Nakashima, 1993; Forsdyke, 1995b), in troponin C genes (Schreier et al. 1990), and, less dramatically, in MHC genes (Milner-White, 1984). This suggests that intron conservation may provide another index of positive selection. The evolution of polymorphic MHC proteins is of much interest (Forsdyke, 1991, 1994, 1995d; Klein and O'hUigin, 1994). While this paper is concerned with positive selection of MHC proteins as part of evolutionary arms races between pathogens and their prey, positive selection may also have been important in the development of a new MHC genes following gene duplications (Goodman, 1975). To become fixed, a new gene would be under pressure to mutate:
There could then be a positive correlation between frequencies of non-synonymous and synonymous mutations in a particular gene (Mouchiroud et al. 1995). This would tend to undermine the value of the ratio of non-synonymous to synonymous mutations as evidence for positive selection. Indeed, quite often this index fails for the peptide-binding domains of polymorphic MHC genes, even though the frequency of non-synonymous mutations in peptide-binding domains is much greater than that in the conserved non-peptide-binding domains (e.g. Ohta, 1995). In this circumstance, detection of decreased stem-loop potential, and some degree of intron conservation, could provide independent supporting evidence for positive selection. Acknowledgement I thank L. Russell for technical help, J. Gerlach for assistance with computer configuration, J. Mau (NRC, Ottawa) for assistance with the Molecular Biology Data Service, and the Medical Research Council of Canada for support.
|
![]()
References
Andersson, G., Larhammer, D., Widmark, E., Servenius, B., Peterson, P. A. and Rask, L. Class II genes of the human major histocompatibility complex. Organization and evolutionary relationship of the DRb genes. J. Biol. Chem. 262: 8748-8758, 1987
Bjorkman, P. J. and Parham, P. Structure, function, and diversity of class I major histocompatibility complex molecules. Annu. Rev. Biochem. 59: 253-258, 1990
Breslauer, K. J., Frank, R., Blocker, H. and Marky, L. A. Predicting DNA duplex stability from base sequence comparison. Proc. Natl. Acad. Sci. USA 83: 3746-3750, 1986
Chorney, M. J., Sawada, I., Gillespie, G. A. Srivastava, R. Pan, J. and Weissman, S. M. Transcription analysis, physical mapping and molecular characterization of a non- classical human leukocyte antigen class I gene. Mol. Cell. Biol. 10: 243-253, 1990
Forsdyke, D. R. Early evolution of MHC polymorphism. J. theor. Biol. 150: 451-456, 1991(Click Here)
Forsdyke, D. R. Relationship of X chromosome dosage compensation to intracellular self/not-self discrimination: a resolution of Muller's paradox? J. theor. Biol. 167: 7-12, 1994
Forsdyke, D. R. A stem-loop kissing model for the initiation of recombination and the origin of introns. Mol. Biol. Evol. 12: 949-958, 1995a
Forsdyke, D. R. Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. An application of FORS-D analysis. Mol. Biol. Evol. 12: 1157-1165, 1995b
Forsdyke, D. R. Relative roles of primary sequence and (G+C)% in determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J. Mol. Evol. 41: 573-581, 1995c
Forsdyke, D. R. Entropy-driven protein self-aggregation as the basis for self/not-self discrimination in the crowded cytosol. J. Biol. Sys. 3: 273-287, 1995d
Forsdyke, D. R. Reciprocal relationship between stem-loop potential and substitution density in retroviral quasispecies under positive Darwinian selection. J. Mol. Evol. 41: 1022-1037,1995d
Forsdyke, D. R. Different biological species "broadcast" their DNAs at different (G+C)% "wavelengths". J. theor. Biol. 178: 405-417,1996a
Girdlestone, J. Nucleotide sequence of an HLA-A1 gene. Nucleic Acids Res. 18: 6701, 1990
Goodman, M., Moore, G. W. and Matsuda, G. Darwinian evolution in the geneology of haemoglobin. Nature 253: 603-608, 1975
Hill, R. E. and Hastie, N. D. Accelerated evolution in the reactive centre regions of serine protease inhibitors. Nature 326: 96-99, 1987
Hughes, A. L. Origin and evolution of HLA class I pseudogenes. Mol. Biol. Evol. 12: 247-258, 1995
Hughes, A. L. and Hughes, M. K. Natural selection on the peptide-binding regions of major histocompatibility complex molecules. Immunogenetics 42: 233-243, 1995
Hughes, A. L. and Nei, M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature 335: 167-170, 1988
Hughes, A. L. and Nei, M. Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proc. Natl. Acad. Sci. USA 86: 958- 962, 1989
Hyldig-Nielsen, J. J., Schenning, L., Hammerling, U., Widmark, E., Heldin, E., Lind, P., Servenius, B., Lund, T., Flavell, R., Lee, J. S., Trowsdale, J., Schreier, P. H., Zablitsky, F., Larhammer, D., Peterson, P. A. and Rask, L. The complete nucleotide sequence of the I-Ead immune response gene. Nucleic Acids Res. 11: 5055-5071, 1983
Jaeger, J. A., Turner, J. H. and Zuker, M. Predicting optimal and suboptimal secondary structure for RNA. Meth. Enzymol. 183: 281-306, 1990
Klein, J. and O'hUigin, C. MHC polymorphism and parasites. Phil. Trans. R. Soc. Lond. B. 346: 351-358, 1994
Koller, B. H., Geraghty, D. E., Shimizu, Y., DeMars, R. and Orr, H. T. HLA-E, a novel class I gene expressed in resting lymphocytes. J. Immunol. 141: 897-904, 1988
Larhammer. D., Hammerling, U., Rask. L. and Peterson, P. A. Sequence of gene and cDNA encoding murine major histocompatibility complex class II gene Ab2. J. Biol. Chem. 260: 14111-14119, 1985
Le, S-Y. and Maizel, J. V. A method for assessing the statistical significance of RNA folding. J. Theor. Biol. 138: 495-510, 1989
Mallissen, M., Hunkapiller, T. and Hood, L. E. Nucleotide sequence of a light chain gene of the mouse I-A subregion: Abd. Science 221: 750-754, 1983
Messer, G., Zemmour, J., Orr, H. T., Parham, P., Weiss, E. H. and Girdlestone, J. HLA- J, a second inactivated class I HLA gene related to HLA-G and HLA-A. J. Immunol. 148: 4043-4053,1992
Milner-White, E. J. Isoenzymes, isoproteins and introns. Trends Biochem. Sci. 9: 517- 519, 1984
Mouchiroud, D., Gautier, C. and Bernardi, G. Frequencies of synonymous substitutions in mammals are gene-specific and correlate with frequencies of non-synonymous substitutions. J. Mol. Evol. 40: 107-113, 1995
Nakashima, K-I., Ogawa, T., Oda, N., Hattori, M., Sakaki, Y., Kihara, H. and Ohno, M. Accelerated evolution of Trimeresurus flavoviridis venom gland phospholipase A2 isoenzymes. Proc. Natl. Acad. Sci. USA 90: 5964-5968, 1993
Nielsen, D. A., Novoradovsky, A. and Goldman, D. SSCP primer design based on single- strand DNA structure predicted by a DNA folding program. Nucleic Acids Res. 23: 2287-2291
Ohta, T. Further examples of evolution by gene duplication revealed through DNA sequence comparisons. Genetics 138: 1331-1337, 1994
Ohta, T. Gene conversion vs point mutation in generating variability at the antigen recognition site of major histocompatibility complex loci. J. Mol. Evol. 41: 115-119, 1995
Ohya, K., Kondo, K. and Mizuno, S. Polymorphism in the human class I MHC locus HLA-E in Japanese. Immunogenetics 32: 205-209, 1990
Radman, M., Wagner, R. and Kricker, M. C. Homologous DNA interactions in the evolution of gene and chromosome structure. Genome Anal. 7: 139-154, 1993
Salter, R. D., Benjamin, R. J., Wesley, P. K., Buxton, S. E., Garrett, T. P., Clayberger, C., Krensky, A. M., Norment, A. M., Littman, D. R. and Parham, P. A binding site for the T cell co-receptor CD8 on the a3 domain of HLA-A2. Nature 345:41-46, 1990
Schamboeck, A., Korman, A. J., Kamb, A. and Strominger, J. L. Organization of the transcriptional unit of a human class II histocompatibility antigen: HLA-DR heavy chain. Nucleic Acids Res. 11: 8663-8675, 1983
Schreier, T., Kedes, L. and Gahlmann, R. Cloning, structural analysis, and expression of the human slow twitch skeletal muscle/cardiac troponin C gene. J. Biol. Chem. 265: 21247-21253, 1990
Scpaer, E. G. and Mullins, J. I. Rate of amino acid change in the envelope protein correlates with pathogenicity of primate lentiviruses. J. Mol. Evol. 37: 57-65, 1993
Sinden, R. R. DNA Structure and Function. Academic Press, San Diego, 1994
Smith, N. H., Maynard Smith, J. and Spratt, B. G. Sequence evolution of the porB gene of Neisseria gonorrhoeae and Neisseria meningitidis: Evidence of positive Darwinian selection. Mol. Biol. Evol. 12: 363-370, 1995
Tanaka,T. and Nei, M. Positive Darwinian selection observed at the variable region genes of immunoglobulins. Mol. Biol. Evol. 6: 447-459, 1989
Zuker, M. (1989). Computer prediction of RNA secondary structure. Meth. Enzym. 180: 262-289.
![]()
Videos on Introns and Exons (Click Here)
Stem-Loop Potential in Retroviruses (Click Here)
Evidence from Overlapping Genes (Click Here)
Introns First 2013 (Click Here)
Return to: Top of Page (Click Here)
Return to: Bioinformatics (Genomics) Index (Click Here)
Go: to Evolution Index (Click Here)
Go: to HomePage (Click Here)
![]()
This page was established circa 1999 and last edited 08 Feb 2013 by Donald Forsdyke