| Deviations from Chargaff's Second Parity Rule Correlate with Direction of Transcription S J. BELL & D. R. FORSDYKEThe Journal of Theoretical Biology (1999b) 197, 63-76 [With permission of the copyright holder, Academic Press] |
 |
 |
|
 |
| Deviations from Chargaff's Second Parity Rule Correlate with Direction of Transcription S J. BELL & D. R. FORSDYKEThe Journal of Theoretical Biology (1999b) 197, 63-76 [With permission of the copyright holder, Academic Press] |
|
|
|
|
2. In E coli Rightward-Transcribing Genes have G-Rich Loop Potential
3. Loop Compositions Reflect Chargaff Differences
4. In W-rich Genomes Rightward-Transcribing Genes have A-Rich Loop Potential
5. Transcription Direction Rule Depends on (C+G)%
6. Transcription Direction Rule in Various Bacterial Species
7. Transcription Direction Rule in Primates
8. R-Rich Clusters in mRNAs Correspond to Loops
9. R-Rich Loops in mRNAs to Avoid Pairing with Other "Self" mRNAs
10. A Mosaic of Loop-Accounting Regions?
| Summary.
The distribution of deviations from Chargaff's second parity rule was examined for overlapping sequence windows of a length (1kb) predicted to be suitable for detecting correlations with functional features of DNA (Bell & Forsdyke 1999a). For long genomic segments from E. coli, Saccharomyces cerevisiae, and Vaccinia virus, Chargaff differences for the W bases and/or for the S bases correlate with transcription direction and gene location. For W-rich genomes, the mRNA-synonymous strand contains regions which, if extruded from negatively supercoiled DNA, would fold to generate stem-loop structures with A-rich loops. Similarly, for S-rich genomes the loops would be G-rich. We suggest that the disposition of genes in nucleic acid sequences arises from their having to adapt to a preexisting mosaic of genomic regions, each distinguished by its potential to extrude single-strand loops enriched for a particular base (or two non-Watson-Crick pairing bases). The mosaic would have facilitated the intrastrand and interstrand accounting required for correction of mutations, and would have evolved in the early RNA world before the emergence of protein-encoding capacity. The preexisting mosaic would have determined transcription direction since there is pressure for all mRNAs of a cell to have purine-rich loops, thus decreasing loop-loop interactions which might lead to formation of "self" sense-antisense RNA duplexes. |
|
Transcribed duplex DNA has an mRNA-synonymous strand and a template strand. If transcription is to the right the top strand is the mRNA-synonymous strand. If transcription is to the left the top strand is the template strand. Three decades ago Szybalski et al. (1966) showed that mRNA-synonymous strands have purine-rich clusters, and Chargaff's famous first parity rule for duplex DNA (%A=%T and %C=%G), was found to apply, to a close approximation, also to single-stranded DNA ("Chargaff' second parity rule"; Karkas et al., 1968; Rudner et al., 1968). Combining Sybalski's observation with Chargaff's second parity rule, it follows for mRNA-synonymous strands, either that purines in the clusters are balanced by an equal number of dispersed pyrimidines, or that there might be small deviations from the second rule ("Chargaff differences") in favour of purines. That such deviation are present, and can act as indicators of transcription direction, is suggested by the above bacterial data from the Chargaff laboratory, by a study of various mammalian genes and viruses by Smithies et al. (1981), and by studies in other organisms (Forsdyke & Bell, 1997; Dang et al., 1998; Cristillo et al., 1998; Bell et al., 1998). Smithies et al. (1981) noted that the "top", mRNA-synonymous, strand of rightward-transcribing globin genes has negative Chargaff differences, when assessed as C-G (i.e. G>C). In the circular SV40 virus genome they noted that negative Chargaff differences in the top strand (i.e. G>C) correlate with the rightward-transcribing late genes (in which the bottom strand is the template strand), and positive Chargaff differences in the top strand (i.e. C>G) correlate with the leftward-transcribing early genes (in which the top strand is the template strand). Thus, as shown for lambdaphage in the prescient Figure 4 of Szybalski et al. (1966), for the "top" strand of DNA, leftward transcription is indicated by pyrimidine excess, and rightward transcription is indicted by purine excess. In the preceding paper we report the sizes of windows in certain long DNA sequences at which average Chargaff differences of the natural sequences differed maximally from those of their shuffled counterparts (Bell & Forsdyke, 1999a). At these window sizes evolutionary forces affecting base order (e.g., generating protein-encoding potential) should be readily detected. We here report the disposition of Chargaff differences in the same sequences, and show that correlations with known sequence features (open reading frames; ORFs) are demonstrated optimally when using window sizes near the predicted optimum. We characterize short-range accounting units, which together with the long-range accounting proposed in the preceding paper, would act to minimize Chargaff differences in long genomic segments. |
2. In E coli Rightward-Transcribing Genes have G-Rich Loop Potential
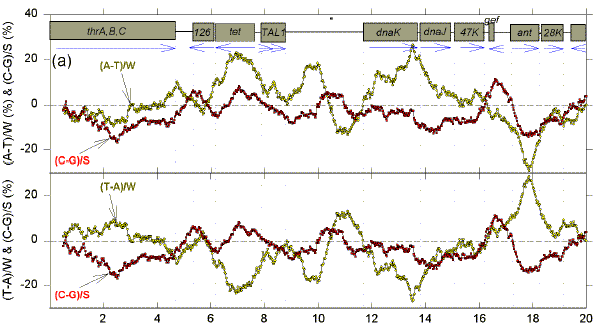
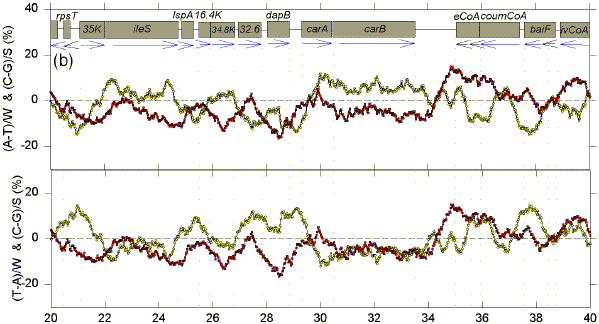
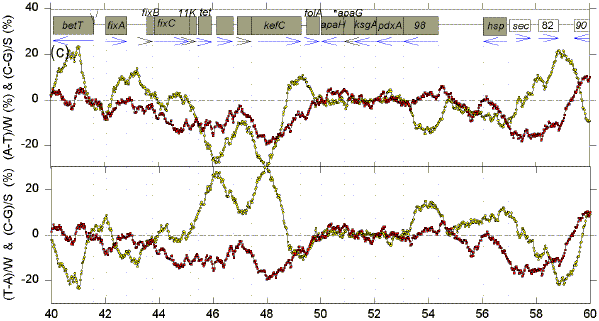
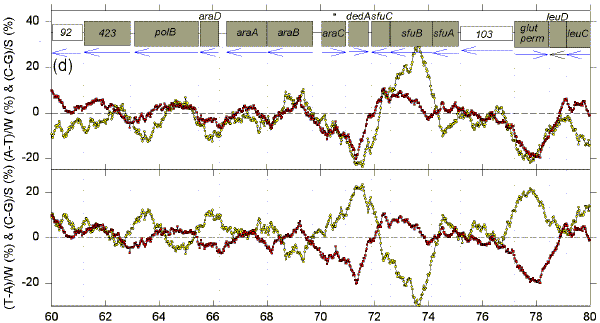
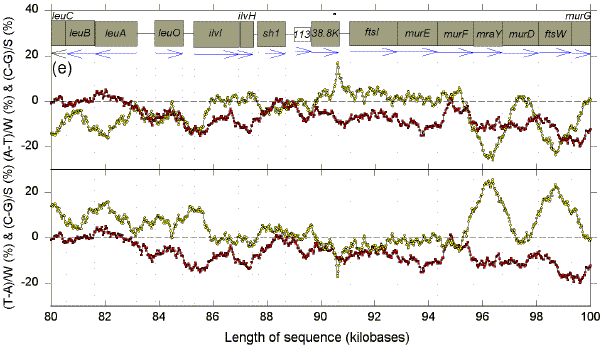
We postulate that genomes are composed of accounting subdomains, which summate to maintain a low overall Chargaff difference (Bell & Forsdyke, 1999a). If a sequence window matches an accounting subdomain (i.e. the centre of the window is near the centre of the domain), then absolute values of the Chargaff differences should be minimal. Fig. 1 shows Chargaff differences for the first 100 kb of GenBank segment ECO110K. A 1 kb window was moved along the sequence in steps of 25 nt, and Chargaff differences were calculated either as (A-T)/W and (C-G)/S (upper figure in each 20 kb section), or as (T-A)/W and (C-G)/S (lower figure in each 20 kb section). In both upper and lower parts of each section of Fig. 1 an excess of Cs over Gs scores positive. In the upper parts an excess of As scores positive. In the lower parts an excess of Ts scores positive (i.e., for both W and S bases an excess of pyrimidines (Y) scores positive, and an excess of purines (R) scores negative). ORFs are shown as boxes. Thus transcription would usually begin close to the left of a box corresponding to a rightward-transcribing gene or operon. |
|
|
|
|
|
|
|
|
|
|
FIG. 1. Variation of Chargaff differences along five 20 kb segments of the first 100 kb of the E. coli sequence ECO110K (a, 0-20 kb; b, 20-40 kb; c, 40-60 kb; d, 60-80 kb; e, 80-100 kb). A 1 kb sequence window was moved in steps of 25 nt and base compositions were determined in each window. Chargaff differences were calculated as described (Bell & Forsdyke, 1999a). Data points are located at the centre of each window. Open yellow circles refer to differences for the W bases, and filled red squares refer to differences for the S bases. The locations of open reading frames (hypothetical and proven) as determined by Yura et al., (1992), are shown as open grey boxes (white boxes are open reading frames with less clearly defined limits). Vertical dotted lines correspond to the limits of open reading frames. Blue horizontal arrows indicate transcription directions assigned to each ORF. |
|
There are regions where W and S Chargaff differences have a common value, which is often close to zero, and regions where the differences have separate values, which often deviate far from zero. The common values are most easily discerned where the curves for the W and S bases cross, and these cross-over regions are sometimes seen better in the upper figure, and sometimes better in the lower figure. The distances between these cross-over regions are variable indicating that, if the regions are located near to the centre of accounting subdomains, then the subdomains are not of uniform size. The postulated centres usually lie in the flanking regions of genes. Thus, in the first 20 kb segment (Fig. 1a, lower), the limits of the leftward-transcribing tetracyclin-resistance gene (tet) are fairly well approximated. Chargaff differences for the W and S bases often show some "synergy", approaching maxima near the centres of genes (which would thus correspond to the borders of the putative accounting subdomains). In Fig. 1a this is particularly evident for the rightward-transcribing ant gene. Thus regions corresponding to genes often appear as curves. Sometimes the curves for the W and S bases project on opposite sides of the zero baseline, and sometimes on the same sides. The accounting subdomains defined at the "micro" level in Fig. 1 might summate to contribute to long range "macro" accounting domains postulated in the previous paper (Bell & Forsdyke, 1999a). Following the series of curves from Fig. 1a to Fig. 1e, it is evident that Chargaff differences for S bases correlate with the direction of transcription. Cs are generally in excess (C>G) when transcription is to the left, and Gs are generally in excess (G>C) when transcription is to the right. The data are summarized in Table 1. For the S bases only 7 of the 29 ORFs transcribed to the left have negative Chargaff differences (i.e., G>C). For the majority (22) of the 29 ORFS C>G (P<0.01). In the case of the W bases, for 23 of the ORFs transcribed to the left T>A; the probability that the average Chargaff difference (involving subtraction of T from A) is less than zero is not significant by Student's t-test (P>0.10), but is significant by the Wilcoxan signed ranks test (P=0.013). For all 40 ORFs transcribed to the right G>C (P<0.001). In the case of the W bases, T>A for 24 of the 40 ORFs; the probability that the average Chargaff difference is less than zero is significant (P<0.01). These data indicate that in E. coli Chargaff differences for the S bases should be more reliable predictors of transcription direction than Chargaff differences for the W bases. Since a minority (7) of 29 leftward transcribing ORFs have G>C, an observation of G>C does not necessarily mean that an ORF is rightward transcribing. However, 40 of the 47 ORFs with G>C are rightward transcribing, so that if the average window in an ORF has G>C this is a good indicator of rightward transcription. On the other hand, no rightward transcribing ORF has C>G, so that if the average window in an ORF has C>G the ORF is very likely to be transcribed to the left. Other genomes also have distinctive Chargaff differences depending on transcription direction (Table 1), as will be discussed later. In general, our observations support the presumption that the 1 kb window size at which Chargaff difference values for natural and shuffled sequences diverge maximally would be the most informative (Bell & Forsdyke, 1998). Thus, the relationships revealed in Fig. 1 were much less apparent when windows smaller or larger than 1 kb were used (e.g. 0.5 kb and 1.5 kb; data not shown). A shuffled sequence gave generally smaller Chargaff differences and patterns which showed no relationship to gene location and transcription direction (data not shown). Plots of differences between non-Watson-Crick base pairs (A-C, G-T, A-G, C-T) sometimes showed relationships to gene location, but not to transcription direction (data not shown). |
3. Loop Compositions Reflect Chargaff Differences
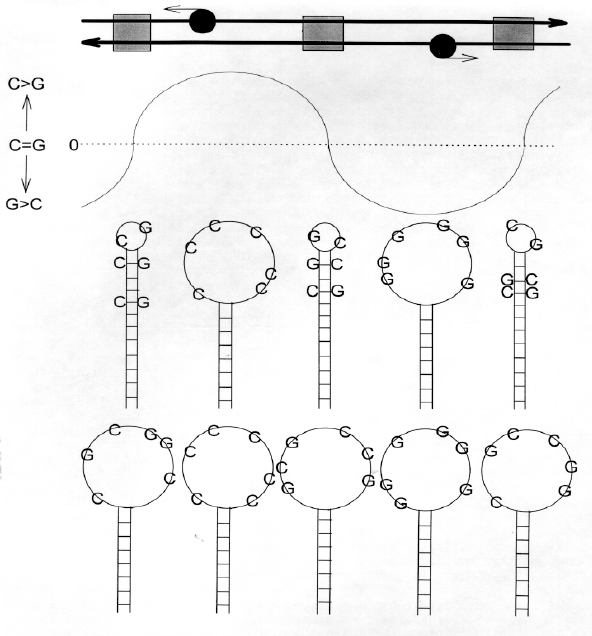
Although the patterns of Chargaff differences around individual genes or groups of genes sometimes show distinctive characteristics (Fig. 1), a general pattern emerges. The upper part of Fig. 2 shows duplex DNA with non-transcribed regions as grey boxes, and leftward- and rightward-transcribing RNA polymerase molecules as black balls. The curves show the relative excess of Cs in the leftward-transcribing region, and of Gs in the rightward-transcribing region. The lower part of the figure shows two possible interpretations of this in terms of stem-loop configurations. |
|
|
FIG. 2. In E. coli the direction of transcription correlates with the relative C- or G-richness of potential loops in the "top" strand. Duplex DNA (long parallel left- and right-pointing arrows representing the upper and lower strands and their 5'->3' polarities) is transcribed by RNA polymerases (black balls), which may utilize as template either upper or lower strands (small arrows indicating direction of transcription), and will transcribe mRNA with the potential for G-rich loops only. Grey boxes refer to non-transcribed DNA containing regulatory elements (e.g., promoters). The curve fluctuating above and below the zero line indicates Chargaff differences (C-G) for the top strand. Potential stem-loop structures which might be extruded from the top strand in supercoiled DNA if base order would allow, are shown as the upper row of stem-loop structures. Large Chargaff differences occurring in the middle of genes confer greater potential for loop formation. The lower row of structures are the corresponding structures if base order will not allow stem formation (i.e. at the ends of genes there is a greater potential for stems since Chargaff differences are low, but base order may not allow stem formation). |
|
On the basis of the overall Chargaff difference values for ECO110K (4.22% for the S bases and 1.24% for the W bases), it is calculated that, on average, up to 97.2% of bases might be paired with their complements in stems. Thus, when there are equivalent numbers of pairing bases, as at the ends of genes (Fig. 1), considerable stem structure might be possible. This is shown in the top row of model stem-loop structures in the lower part of Fig. 2. However, although the correct bases are present in the correct proportions (i.e., base composition is compatible with extensive stem formation), base order may not permit this. This is shown in the bottom row of model stem-loop structures shown in the lower part of Fig. 2. To distinguish between these possibilities, 15 sequence windows corresponding to a wide range of Chargaff difference values were selected from those shown in figure 1. The 1 kb sequences were each subjected to the energy-minimization folding program LRNA (Jaeger et al., 1990), using energy parameters suitable for folding DNA at 37oC (SantaLucia et al., 1996). On average, 57.8% (standard error �0.4%) of bases were in stems. Although windows with small Chargaff differences would seem to have greater potential for stem formation, this was not observed in folded structures. The magnitude of Chargaff differences was neither positively correlated with the proportion of bases in loops (whether a "loop" is loosely defined as any non-pairing base, or >4 non-pairing bases), nor inversely correlated with the proportion of bases in stems. Thus base order plays an important role in determining calculated structure. Since in stems Chargaff differences tend to be zero (by definition), then overall Chargaff differences should be reflective of the base composition of loops. Indeed, as expected, Chargaff differences calculated directly from the base composition of loops correlated well with Chargaff differences calculated for the corresponding sequence windows (r=0.98). Thus Chargaff differences (i.e., relative richness of a region for a particular W base or S base) would be reflected in the composition of loops in the stem-loop structures which might be extruded from supercoiled DNA under biological conditions (Murchie et al., 1992). These folding studies were repeated with smaller windows (200 nt), with similar results. A study of a human genomic segment HUMMMDBC using 200 nt windows also produced similar results (data not shown). |
| Chargaff difference formula | (A-T)/W (%) | (C-G)/S (%) | |||
| Direction of transcription | To Left | To Right | To Left | To Right | |
| DNA sourceb | (C+G) % | . | . | ||
| E. coli | 52.6 | T>A 23:6 (P=0.013) -1.83+1.3 (P>0.10) |
T>A 24:16 (P=0.011) -3.86+1.3 (P<0.01) |
C>G 22:7 (P=0.007) 1.64+0.6 (P<0.01) |
G>C 40:0 (P<0.00003) -7.71+0.6 (P<0.001) |
| S. cerevisiae III | 38.5 | T>A 70:30(P=0.001) -2.80+1.0 (P<0.005) |
A>T 64:14(P<0.00003) 5.29+1.0(P<0.0005) |
C>G 70:30 (P=0.0014) 2.64+0.9 (P<0.005) |
C>G 42:36 (P=0.15) 0.90+1.0 (P>0.10) |
| Vaccinia virus | 33.4 | T>A 94:11 (P<0.0003) -4.8+0.4(P<0.0005) |
A>T
|
C>G 75:30(P<0.00003) 4.26+0.8(P<0.0005) |
G>C 70:22(P<0.00003) -5.22+0.7(P<0.0005) |
| a Chargaff difference values for an individual ORF
were determined as the average of the Chargaff differences of all the 1 kb windows whose
centres were within the ORF. The second row of data shows the relative proportions of ORFs with positive and negative Chargaff differences, together with probabilities that the asymmetries in numbers of negatives and positives are not significant (calculated using the Wilcoxan signed ranks test). |
|||||
4. In W-rich Genomes Rightward-Transcribing Genes have A-Rich Loop Potential
The study shown in Fig. 1 for E. coli segment ECO110K (C+G=52.8%), was repeated for the W-rich Vaccinia virus genome (C+G=33.4%; Goebel et al., 1990), and for chromosome III of Saccharomyces cerevisiae (C+G=38.5%; Oliver et al., 1992). Again, correlations with transcription direction were obvious in the natural sequence, but not in a shuffled version (data not shown). In the case of Vaccinia virus, Chargaff differences for the W bases correlate best with transcription direction (A>T in 83 out of 92 rightward transcribing ORFs), but S bases are also often good predictors (Table. 1). In the case of chromosome III of Saccharomyces cerevisiae, Chargaff differences for the W bases correlate well with transcription direction, but Chargaff differences for the S bases are less reliable. A study of the bithorax region from the W-rich genome of Drosophila melanogaster showed that A>T in the case of rightward transcribing genes and ORFs (Dang et al., 1998). |
| DNA sourcea | (C+G)% | Bases predicting rightward ORFs by Chargaff differences | Reference |
| C. perfringens | 31 | A>T (G>C)b | Szybalski et al. 1966 |
| Vaccinia virus (VACCG) | 33.4 | A>T (G>C) | This paper |
| T2 phage | 34 | A>T | Szybalski et al. 1966 |
| Herpes saimiri | 35 | A>T | Cristillo et al. 1998 |
| S. cerevisae III | 38.5 | A>T | This paper |
| Simian foamy virus-1 | 39.2 | A>T (G>C) | Cristillo et al. 1998 |
| Fetal globin (human) | 39.5 | G>C | Smithies et al. 1981 |
| Beta-globin (mouse) | 39.8 | T>A | Smithies et al. 1981 |
| Simian virus 40 | 40.8 | G>C | Smithies et al. 1981 |
| Drosophila bithorax | 41.7 | A>T | Dang et al. 1998 |
| HIV-1 | 42.6 | A>T (G>C) | Cristillo et al. 1998 |
| Varicella-Zoster virus | 46.1 | A>T | Cristillo et al. 1998 |
| Polyoma virus A-2 | 47.2 | A>T | Smithies et al. 1981 |
| Lambdaphage | 50 | G>C | Szybalski et al. 1966 |
| T7 phage | 50 | G>C | Szybalski et al. 1966 |
| B. subtilis | 50 | G>C | Szybalski et al. 1966 |
| E. coli (ECO110K) | 52.6 | G>C | This paper |
| HTLV-1 | 53.2 | C>G | Cristillo et al. 1998 |
| Rous sarcoma virus | 54.4 | G>C (A>T) | Cristillo et al. 1998 |
| Epstein-Barr virus | 60.1 | C>G | Cristillo et al. 1998 |
| Herpes simplex (HE1CG) | 68.3 | C>G | Cristillo et al. 1998 |
| M. lysodeikticus | 71 | G>C | Szybalski et al. 1966 |
| S. lutea | 71 | G>C | Szybalski et al. 1966 |
| a Genbank names are capitalized and contained in parentheses. b Parentheses indicate a minor role. | |||
5. Transcription Direction Rule Depends on (C+G)%
Table 2 summarizes our data and some data from the early literature, which can now be reinterpreted. The top strand for rightward-transcribed ORFs usually has R-rich loops. At low C+G percentages the main predictor of rightward transcription is A, either alone or accompanied by G. At high C+G percentages G is the main purine predictor. Thus, results from various species (Tables 1, 2) strongly support what might be referred to either as the "Chargaff difference transcription rule", or as "Szybalski's transcription direction rule" (Szybalski et al., 1966). Rather than question the validity of the rule, when individual genes/ORFs are found within a genome or genome sector which disobey the rule for that genome or genome sector (Tables 1, 2), it is appropriate to ask whether those genes/ORFs have special characteristics, or are misassigned or non-functional (Dang et al., 1998). However, there are also exceptional genomes which disobey the rule. These are the S-rich genomes of the human T cell leukaemia virus (HTLV-1), Herpes simplex virus-1, and Epstein-Barr virus (Table 2). These exceptions are all viruses which are profoundly committed to latency, in which state only a few mRNAs are expressed. Remarkably, latency-associated mRNAs in the viruses tend to be exceptional and obey the rule (Cristillo et al., 1998). |
|
|
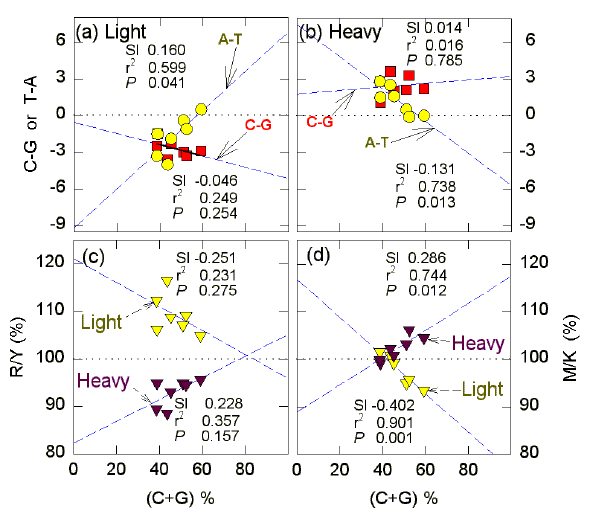
FIG. 3. Relationships between (C+G)% of different bacterial species and genomic Chargaff ratios, as determined by Rudner et al. (1969). Chargaff differences were calculated directly from the chemically-determined base compositions (mole %) of "light" and "heavy" strands (e.g. for the ordinate of Fig. 3a, "T-A" refers to the mole % T less the mole % of A). Genomes were from B. megaterium (38.6% C+G), P. vulgaris (38.8% C+G), B. subtilis (43.5% C+G), B. stearothermophilus (45.2% C+G), E. coli (51.0% C+G), S. typhimurium (52.3% C+G), and S. marcescens (59.2% C+G). In (a) and (b) points for the W bases are with open yellow symbols, and points for the S bases are with filled red symbols. In (c) and (d) points for the light strands are with open yellow symbols, and points for the heavy strands are with filled symbols. Parameters of the least squares regression plots (dashed lines) are in blocks near to the relevant line; Sl corresponds to the slope; r2 is the square of the correlation coefficient (unadjusted); P is the probability that the slope is not significantly different from zero. ("Light" and "heavy" are operational terms used when separating DNA strands chromatographically or by ultracentrifugation; purine-rich strands tend to be "lighter" than the "heavy" pyrimidine-rich strands). |
6. Transcription Direction Rule in Various Bacterial Species
Further evidence on the role of base composition is found in data gathered by Rudner et al. (1969) from the genomes of several bacterial species which span a wide range of C+G percentages (Fig. 3). At low (C+G)%, both purines contribute to equal extents to average Chargaff differences in the light (mRNA synonymous) strands (Fig. 3a). As (C+G)% increases the contribution of the W bases to average Chargaff differences for the genomes decreases to near zero, whereas the contribution of the S bases remains constant, or may increase slightly (but not significantly in this study; P >0.05). Reciprocal changes are noted with the "heavy" template strand, where both pyrimidines contribute to Chargaff differences at low (C+G)% (Fig. 3b). Average purine/pyrimidine ratios in the light and heavy strand sequences do not change significantly with increasing (C+G)%, although there are suggestive trends (Fig. 3c). However, the average ratios between the 6-amino (A, C) and 6-keto (G, T) bases show significant changes (Fig. 3d). At 40% (C+G), when both purines contribute to the R-richness of the mRNA-synonymous strand, the 6-amino and 6-keto bases are in balanced proportions; but as C+G percentages increase the light, mRNA-synonymous, strand loses more As than Ts, and increases Gs more than Cs. Thus, the denominator increases with respect to both 6-keto bases, and the numerator decreases with respect to both 6-amino bases. Reciprocal changes are seen in the heavy (template) DNA strand. |
7. Transcription Direction Rule in Primates
Our results are consistent with the recent generalization of Mrazek and Kypr (1994) that A>T universally in protein-encoding sequences (from bacteria to primates). From their data on 3,954 primate coding sequences, the average Chargaff difference for the W bases (calculated as (A-T)/W and expressed as a percentage), is 8.4%. Similarly, the base composition of 1657 human coding sequences compiled by Karlin & Mrazek (1996), yields a generic Chargaff difference (calculated as (R-Y)/(R+Y) and expressed as a percentage) of 4%. |
8. R-Rich Clusters in mRNAs Correspond to Loops
|
Although the energy parameters for folding RNA differ from those for folding DNA (SantaLucia et al., 1996), there are usually not major differences between the structure generated by folding a mRNA and the structure generated by folding its cognate mRNA-synonymous DNA strand (D. R. Forsdyke, unpublished work). Thus, our results for mRNA-synonymous strands should apply to the corresponding mRNAs. The loop regions of the mRNAs of various species should be preferentially enriched in certain bases. For E. coli, the loops would be enriched for G (Fig. 2). The studies of Eguchi et al. (1991) indicate that loop-loop "kissing" interactions are important in initiating hybridization between nucleic acids (see later). Thus, our results are consistent with the observation of Szybalski et al. (1966) that the template strand of B. subtilis DNA rapidly hybridizes at low temperature (4-25�) with poly rG to form partial double-strand complexes. (At higher temperatures the template strand would slowly hybridize with bacterial mRNA to form perfect complexes.) The rapid low-temperature hybridization was ascribed to C-rich "clusters" in the template strand; this implied that there were G-rich "clusters" in the mRNA-synonymous strand of the DNA. It should be noted that if an mRNA (and hence the corresponding mRNA- synonymous strand) has purine clusters without complementary pyrimidine clusters in close proximity, then a priori the most likely location of the purines would be in the loops of any stem-loop secondary structures adopted by the mRNA (or mRNA-synonymous strand). It was also observed that sheared, denatured, B. subtilis DNA could be separated into "light" and "heavy" fractions, considered to represent the two complementary strands of the native duplex (Karkas et al., 1968; Rudner et al., 1968). These could be transcribed by E. coli RNA polymerase in vitro to generate RNA products with complementary base compositions. The light fraction generated a Y-rich product, and was itself R-rich; the heavy fraction generated an R-rich product, and was itself Y-rich. However, when native DNA was used as template, the quantity of product was half that produced by the denatured strands, and its composition was similar to that produced by the heavy fraction, being particular rich in G. Thus, in the absence of other proteins, the RNA polymerase appeared able to monitor duplex DNA with respect to transcription direction. This implies that the polymerase monitors some sequence feature. |
9. R-Rich Loops in mRNAs to Avoid Pairing with Other "Self" mRNAs
|
Why should the majority of cell mRNAs have the potential to form loops enriched in the same base? Although natural mRNAs ("sense mRNAs") are likely to exist as partial ribonucleoprotein complexes within cells, the corresponding antisense mRNAs introduced experimentally either by injection (Melton, 1985), or by transcription from transfected artificial constructs (Izant & Weintraub, 1984), are able to inhibit expression of the natural mRNAs with high specificity (Krol et al., 1988). This indicates that at least some parts of natural sense RNAs are available for hybridization to antisense RNAs. Indeed, there are many examples of natural "antisense" RNAs, which control with high specificity the expression of the transcript from the complementary strand (the "sense" mRNA; Eguchi et al., 1991). The specificity of these artificial or natural RNA-RNA interactions is determined by the precise complementarity of pairing strands. However the pairing is initiated by reversible exploratory interactions between the tips of the loops of stem-loop structures (Eguchi et al., 1991; Marino et al., 1996). These initial, reversible, "kissing" interactions are relatively non-specific, but provide an alignment between two strands, which may be followed either by full hybridization, or by dissociation of the complex. A similar mechanism has been suggested as a basis for the pairing between homologous DNA strands required for mitotic or meiotic recombination (Kleckner & Weiner, 1993; Kleckner, 1997); one function of this would be the detection and correction of sequence errors (discussed below). The intracellular environment should be highly adapted to favour exploratory, low-specificity, loop-loop interactions between nucleic acids. An obvious example of this would be the interactions between anticodon loops of tRNA molecules, and codons in mRNA. Protein synthesis occurs extremely rapidly, even though for each amino acid added to the growing peptide chain there must be discrimination between many competing tRNA species. It is likely that the "crowded" cytosol provides an environment where such recognition reactions, probably with a high entropy-driven component, can occur very efficiently (Lauffer 1975; Cantor & Schimmal, 1980; Forsdyke 1995; Zimmerman & Murphy, 1996). Given such a favourable environment for initial low-specificity loop-loop interactions of an exploratory nature between RNA molecules, there should have been an evolutionary pressure to avoid unnecessary loop-loop interactions, such as between "self" mRNA molecules which happened to have complementary loops. In a crowded cytoplasm where mRNAs with G-rich loops predominated, there would be little likelihood of a G-rich mRNA promiscuously pairing with one of its fellows. A gene encoding a minority mRNA population with C-rich loops would be at a strong selective disadvantage since the mRNA molecules would be diverted by multiple transient interactions with members of the G-rich loop-bearing majority species. However, if at an important time in the life of a cell only a limited number of specific mRNAs predominated (e.g., in reticulocytes synthesizing mainly globin), there would then be the possibility of adopting a loop pattern in response to other selective pressures (e.g., facilitating codons for abundant tRNAs), rather than to the pressure of having to have a loop pattern adapted to avoid ephemeral "kissing" with other mRNA species. In this respect it is of interest to note that a mouse b-globin mRNA appears exceptional in having high Y-rich loop potential (Table 2).In organisms with intron-containing genes, mRNAs are generated by splicing short-lived primary transcripts. The latter are located in the nucleus at low concentrations, so that there would be less need to have purine-rich loops in introns to avoid "kissing" interactions with other RNAs. This would predicts that introns would not show the purine bias, as has indeed been noted (Mrazek and Kypr, 1994). |
10. A Mosaic of Loop-Accounting Regions?
|
Deviations from Chargaff's second rule are manifest as a series of curves which demarcate genes or groups of genes sharing a common direction of transcription (Figs 1, 2). In E. coli some of the groups could correspond to operons, where several gene products are generated from a polycistronic mRNA. However, similar group demarcations are seen in organisms which do not have polycistronic mRNA (e.g., Saccharomyces cerevisiae; data not shown). The clustering of yeast genes sharing a common transcription direction does not appear to have arisen on a random basis (Bussey et al., 1995). It has been proposed that stem-loop potential developed in the early "RNA world" to assist intrastrand and interstrand (recombination) repair processes (Forsdyke, 1996). The need for a complementary base would have allowed detection of base mutations, which would have been manifest through base mispairing (Bernstein & Bernstein, 1991). Intrastrand accounting would have involved pairing interactions between bases in the complementary stems and loops of stem-loop structures. Kleckner has suggested (Kleckner & Weiner, 1993; Kleckner, 1997) that pairing interactions between bases in complementary loops could also be important for initiation of interstrand pairing in meiosis (which would facilitate recombination repair), a process which might remain localized without the exchange of distant markers (Bowring & Catcheside, 1996). This primitive system would have persisted in the subsequent early "DNA world". Loops would have had the same complementarity requirements as stems and, since loop-loop interactions would have tended to be over a longer range, sequence modifications which might have accelerated the sequence search process would have been selected for. Thus a region with C-rich loops might initially have contacted a region with G-rich loops through pairing ("kissing") between incorrect Cs and Gs. Having established contact, realignment of strands would have allowed exploration of the possibility of a better alignment, or rejection of the interaction (Eguchi et al., 1991). A tendency to polarize towards becoming either C-rich or G-rich (for example), to accelerate the initial "kissing", would have been selected for. This hypothesis would predict the evolution of a genome consisting of a mosaic of accounting regions with distinctive complementary loops. For example, regions with loops rich in A and C with A>C (e.g. tet in Fig. 1a), might be accounted for elsewhere in the genome by regions with loops rich in T and G with T>G (e.g. ant in Fig. 1a). How one arm of a cruciform with its stem-loops, while seeking a pairing partner elsewhere, might evade its complementary arm extruded from the other strand, is a biochemical problem we cannot usefully discuss at this time.
|
At some stage the early "replicators" began to "build" themselves "survival machines" (Dawkins 1989). Protein-encoding capacity would have been imposed on genomes already highly adapted for stem-loop formation. Determined by some initial restrictive base or sequence requirement for the direction of transcription (such as the need for R-rich loops in all mRNAs; see above), the pre-existing mosaic would have determined in which directions the first protein-encoding genes were transcribed (Fig. 2). Thus, the direction of transcription would have been determined by the mosaic, and not the converse. Cross-over regions (from Y-richness to R-richness, and vice-versa) would have defined the limits of the initial transcription units. |
|
A linkage between transcription and recombination has been suggested many times (Cook, 1997; Nicolas, 1998). Support for this arises from recent studies of the "cross-over hotspot instigator" (Chi) sequence in E. coli. The domain of the eight base Chi sequence has been found to extend to approximately 0.8 kb (Tracy et al., 1997), which closely corresponds to the window size which we find optimum (Bell & Forsdyke, 1998). Furthermore, Chi sequences in the top strand are usually in rightward transcribing ORFs, whereas Chi sequences in the bottom strand are in leftward-transcribing ORFs (Bell et al., 1998). We have here indicated the size of "micro" accounting units operating at the level of individual genes or gene groups. These would work within the context of long range "macro" accounting units proposed in the preceding paper (Bell & Forsdyke, 1998) to decrease deviations from Chargaff's second rule. Cooperation between short- range and long-range accounting processes might be mechanistically quite complex (Forsdyke, 1981). We note that the limits of short-range accounting units, defined as the regions where Chargaff differences reach maxima, tend to correspond to the middle of genes (Figs 1, 2). The middle of short-range accounting units corresponds to the end of genes. On the other hand, long-range "kissing" interactions can be presumed to begin in the middle of the curves which demarcate genes or gene groups, and to terminate at the cross-over regions associated with the ends of genes or gene groups. Thus, long-range accounting units correspond to transcriptional units. Short-range and long-range accounting units would appear to overlap. |
11. Mutation with Strand Bias?
Using arbitrary window sizes, Lobry (1996a, b) noted that Chargaff differences can be used to identify origins of replication in Mycoplasma genitalium, and in some other bacteria. Chargaff differences for C-G are positive (C>G) to the "left" of replication origins, and negative (G>C) to the "right" of replication origins. Lobry suggested a mutational bias resulting from mutation differences between the leading and lagging strands at the replication fork. Strand biases in mutations might also reflect differences in repair processes, which appear to affect differentially the transcribed (template) strands and the non-transcribed (mRNA-synonymous) strands (Hanawalt, 1991). Thus, an alternative explanation, consistent with the mutational bias viewpoint, would be that genes close to the replication origin are transcribed to the left to the "left" of the replication origin, and transcribed to the right to the "right" of the replication origin. This is indeed found in Mycoplasma genitalium (Fraser et al., 1995), as in SV40 virus (Smithies et al., 1981). However, the neutralist view that mutational biases play a major role in genetic phenomena (Ohta, 1996), is controversial (Bernardi, 1993; Bernardi et al., 1993; Forsdyke 1996, 1998). We have suggested here that selective forces have been dominant. While appealing to the parsimoniously inclined, a purely stochastic explanation for Chargaff's second parity rule (Lobry, 1995) seems unlikely. |
We thank P. Sibbald for review of the manuscript, J. Gerlach for assistance with computer configuration, L. Russell for technical help, J. SantaLucia for making available parameters for DNA for use in the folding program LRNA, R. Gough, J. Mau and G. Wood for facilitating use of Genetics Computer Group software on the Silicon Graphics computer at the National Research Council, Ottawa, and T. Smith for statistical advice. The work was supported by the Medical Research Council of Canada and Queen's University.
BELL, S. J., CHOW, Y. C., HO, J. Y. K. & FORSDYKE, D. R. (1998). Correlation of CHI orientation with transcription indicates a fundamental relationship between recombination and transcription. Gene 216, 285-292.
BELL, S. J. & FORSDYKE, D. R. (1999a). Accounting units in DNA. J. theor. Biol. 197, 51-61.
BERNARDI, G. (1993). The vertebrate genome: isochores and evolution. Mol. Biol. Evol. 10, 186-204.
BERNARDI, G., MOUCHIROUD, D. & GAUTIER, C. (1993). Silent substitutions in mammalian genomes and their evolutionary implications. J. Mol. Evol. 37, 583- 589.
BERNSTEIN, C. & BERNSTEIN, H. (1991). Aging, Sex and DNA Repair. San Diego, Ca: Academic Press.
BOWRING, F. J. & CATCHESIDE, D. E. A. (1996). Gene conversion alone accounts for more than 90% of recombination events at the am locus of Neurospora crassa. Genetics 143, 129-136.
BUSSEY, H. et al., (1995). The nucleotide sequence of chromosome I from Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 92, 3809-3813.
CANTOR, C. R. & SCHIMMEL, P. R. (1980). Biophysical Chemistry. pp. 1183-1264. San Francisco, CA: W. H. Freeman and Co.
COOK, P. R. (1997). The transcriptional basis of chromosome pairing. J. Cell Sci. 110, 1033-1040.
CRISTILLO, A. D., LILLICRAP, T. P. & FORSDYKE, D. R. (1998). Purine- loading of EBNA-1 mRNA avoids sense-antisense "collisions". FASEB. J. 12, A1453.
DANG, K. D., DUTT, P. B. & FORSDYKE, D. R. (1998). Chargaff differences correlate with transcription direction in the bithorax complex of Drosophila. Biochem. Cell Biol. 76, 129-137.
DAWKINS. R. (1989). The Selfish Gene. New York, NY: Oxford University Press.
EGUCHI, Y., ITOH, T. & TOMIZAWA, J. (1991). Antisense RNA. Annu. Rev. Biochem. 60, 631-652.
FORSDYKE, D. R. (1995). Entropy-driven protein self-aggregation as the basis for self/not-self discrimination in the crowded cytosol. J. Biol. Sys. 3, 273-287.
FORSDYKE, D. R. (1996). Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths". J. theor. Biol. 178, 405-417.
FORSDYKE, D. R. (1999) The origin of species, revisited. Queen's Quarterly 106, 110-132.
FORSDYKE, D. R. & BELL, S. J. (1997). Deviations from Chargaff's second rule correlate with direction of transcription and genome structure. Proc. Can. Fed. Biol. Socs. 40, 87.
FRASER, C. M. et al., (1995). The minimal gene complement of Mycoplasma genitalium. Science 270, 397-403.
GOEBEL, S. J., JOHNSON, G. P., PERKUS, M. E., DAVIS, S. W., WINSLOW, J. P. & PAOLETTI, J. (1990). The complete DNA sequence of vaccinia virus. Virology 179, 247-266.
HANAWALT, P. C. (1991). Heterogeneity of DNA repair at the gene level. Mut. Res. 247, 203-211.
IZANT, J. G. & WEINTRAUB, H. (1984). Inhibition of thymidine kinase gene expression by anti-sense RNA: a molecular approach to genetic analysis. Cell 36, 1007-1015.
JAEGER, J. A., TURNER, D. H. & ZUKER, M. (1990). Predicting optimal and suboptimal secondary structure for RNA. Meth. Enzymol. 183, 281-306.
KARKAS, J. D., RUDNER, R. & CHARGAFF, E. (1968). Separation of B. subtilis DNA into complementary strands, II. Template functions and composition as determined by transcription with RNA polymerase. Proc. Natl. Acad. Sci. USA 60, 915-920.
KARLIN, S. & MRAZEK, J. (1996). What drives codon choice in human genes? J. Mol. Biol. 262, 459-471.
KLECKNER, N. (1997). Interactions between and along chromosomes during meiosis. Harvey Lectures 91:21-45
KLECKNER, N. & WEINER, B. M. (1993). Potential advantages of unstable interactions for pairing of chromosomes in meiotic, somatic and premeiotic cells. Cold Spring Harbor Symp. Quant. Biol. 58, 553-565.
KROL, A. R., VAN DER MOL, J. N. M. & STUITJE, A. R. (1988). Modulation of eukaryotic gene expression by complementary RNA or DNA sequences. Biotechniques 6, 958-976.
LAUFFER, M. A. (1975). Entropy-driven Processes in Biology. New York, NY: Springer-Verlag.
LOBRY, J. R. (1995). Properties of a general model of DNA evolution under no- strand-bias conditions. J. Mol. Evol. 40, 326-330.
LOBRY, J. R. (1996a). Asymmetric substitution patterns in the two DNA strands of bacteria. Mol. Biol. Evol. 13, 660-665.
LOBRY, J. R. (1996b). Origin of replication of Mycoplasma genitalium. Science 272, 745-746.
MARINO, J. P., GREGORIAN, R. S., CSANKOVSZKI, G. & CROTHERS, D. M. (1996). Bent helix formation between RNA hairpins with complementary loops. Science 268, 1448-1454.
MELTON, D. A. (1985). Injected anti-sense RNAs specifically block messenger RNA translation in vivo. Proc. Natl. Acad. Sci. USA 82, 144-148.
MRAZEK, J. & KYPR, J. (1994). Biased distribution of adenine and thymine in gene nucleotide sequences. J. Mol. Evol. 39, 439-447.
MURCHIE, A. I. H., BOWATER, R., ABOUL-ELA, F. & LILLEY, D. M. J. (1992). Helix opening transitions in supercoiled DNA. Biochem. Biophys. Acta 1131, 1-15.
NICOLAS, A. (1998). Relationship between transcription and initiation of meiotic recombination: towards chromatin accessibility. Proc. Natl. Acad. Sci. USA 95, 87- 89.
OHTA, T. (1996). The current significance and standing of the neutral and near-neutral theories. BioEssays 18, 673-684.
OLIVER, S. G., et al., (1992). The complete DNA sequence of yeast chromosome III. Nature 357, 38-46.
RUDNER, R, KARKAS, J. D. & CHARGAFF, E. (1968). Separation of B. subtilis DNA into complementary strands, III. Direct analysis. Proc. Natl. Acad. Sci. USA 60, 921-922.
RUDNER, R., KARKAS, J. D. & CHARGAFF, E. (1969). Separation of microbial deoxyribonucleic acids into complementary strands. Proc. Natl. Acad. Sci. USA 63, 152-159.
SANTALUCIA, J., ALLAWA, H. T. & SENEVIRATNE, P. A. (1996). Improved nearest neighbour parameters for predicting DNA duplex stability. Biochemistry 35, 3555-3562.
SMITHIES, O., ENGELS, W. R., DEVEREUX, J. R., SLIGHTOM, J. L. & SHEN, S. (1981). Base substitutions, length differences and DNA strand asymmetries in the human G
l and Al fetal globin gene region. Cell 26, 345-353.SZYBALSKI, W., KUBINSKI, H. & SHELDRICK, P. (1966). Pyrimidine clusters on the transcribing strands of DNA and their possible role in the initiation of RNA synthesis. Cold Spring Harbor Symp. Quant. Biol. 31, 123-127.
TRACY, R. B., CHEDIN, F. & KOWALCZYKOWSKI, S. C. (1997). The recombinational hot-spot Chi is embedded within islands of preferred DNA pairing sequences in the E. coli genome. Cell 90, 205-206
YURA, T., MORI, H., NAGAI, H., NAGATA, T., ISHIHAMA. A., FIJITA, N., ISONO, K., MIZOBUCHI, K. & NAKATA, A. (1992). Systematic sequencing of the Escherichia coli genome: analysis of the 0-2.4 min region. Nucleic Acids Res. 20, 3305-3308.
ZIMMERMAN, S. B. & MURPHY, L. D. (1996). Macromolecular crowding and the mandatory condensation of DNA in bacteria. FEBS Lett. 390, 245-248.
Return: to Accounting Units in DNA (Bell & Forsdyke 1999a) (Click Here)
Return: to HomePage (Click Here)
Return: to Bioinformatics (Genomics) Index (Click Here)
This page was established circa 1999 and was lasted edited on 04 Apr 2009 by Donald Forsdyke