To build a house you need bricks and mortar and something else -- the "know-how", or "information" as to how to go about your business. The Victorians knew this. But when it came to the "building" of animals and plants, -- the word, and even the concept, of "information" is difficult to trace in their writings. Classical scholars tell us that Aristotle did not have this problem. The "eidos", the form-giving essense that shapes the embryo "contributes nothing to the the material body of the embryo but only communicates its program of development" (see Delbruck's "Aristotle-totle-totle" in Of Microbes and Life 1971, pp. 50-55). William Bateson spoke of a "factor" (gene) having the "power" to bring about the building of the characters which make up an organism (Click Here). He used the "information" concept, but not the word. He was prepared to believe his factors were molecules of the type we would today call macromolecules, but he did not actually call them "informational macromolecules". As set out elsewhere in these pages (Click Here) the story, beginning in the 1870s with Ewald Hering in Prague and Samuel Butler in London, and proceeding via Richard Semon to Erwin Schrodinger (Click Here), has escaped modern commentators (e.g. Schrodinger's What is Life? 1944; L. E. Kay 1995. Science in Context 8, 609-634). But certainly by the time of the discovery of the double helical structure of DNA in 1953 the concept was established (see Olby's The Path to the Double Helix, 1974, and Portugal and Cohens' A Century of DNA, 1977). |
 |
 |
Information has many forms. If you turn down the corner of a page of a book to remind you where you stopped reading ("book-mark"), then you have left information on the page. In future you read ("decode") the bookmark with the knowledge that it means "continue here". A future historian might be interested in where you paused in your reading. Coming across the book, he/she would notice creases suggesting that a flap had been turned down. Making assumptions about the code you were using, a feasible map of the book could then be made with your pause sites. It might be discovered that you paused at particular sites, say at the ends of chapters. In this case pauses would be correlated with the distribution of the book's "primary information". Or perhaps there was a random element to your pausing ... perhaps when your partner wanted the light out. In this case pausing would be influenced by your pairing relationship. A more familiar form of information is the linear form you are now decoding (reading), which is similar to the form you might decode on the page of a book. If a turned-down flap on a page is a large one, it might cover up some of the information. Thus, one form of information might interfere with another form of information. To read the text you would have to correct (fold back) the "secondary structure" of the page (the flap) so that it no longer overlapped the text. Thus, there is a conflict. You can either retain the flap and not read the text, or get rid of the flap and read the text. In the case of a book page, the text is imposed on an underlying flat two dimensional base, the paper. Similarly, in the case of our genetic material, DNA, the "paper" is a chain of two units (phosphate and ribose) and the most easily recognized "text" is provided by a sequence of "letters" (bases) attached to the chain. As in the case of a written text on paper, "flaps" in DNA (secondary structure) can conflict with the base sequence (primary structure). Thus the pressures to convey information encoded in a particular sequence, and to convey information encoded in a "flap", may be in conflict. The "hand" of evolution has to resolve these apparently intrinsic conflicts while dealing with other pressures (extrinsic) from the environment. The stunning novelty of the Watson-Crick model of DNA was not only that it was beautiful, but that it also explained so much of the biology of heredity. There was not just one sequence of letters, but two. These were wrapped round each other in the form of a double helix. One was the complement of the other, so that the sequence of one string (strand) could be inferred from the sequence of the other. If there were damage to one strand of DNA, then that strand could potentially be repaired on the basis of the text of the opposite strand. When the cell divided the two strands would part and separate. New "child" strands, synthesized from nucleotide "building blocks" (each consisting of phosphate, ribose and a base) , would replace those which had separated, so that duplexes identical to the parental duplex would be created. There were two main types of bases, purines (R) and pyrimidines (Y). Thus, disregarding the phosphate-ribose chain, the first nucleic acids to appear in evolution could accurately be represented as a binary sequence such as RYRRYRYYRYR.... Each base would be a "binary digit". Conventionally, we represent binary digits in computer language as strings of 0s and 1s. If a Y and an R were equally likely alternatives in a sequence position, then each could be quantitated as one "bit" of information. Each main type of base came in two flavours. Two types of purines, adenine (A) and guanine (G), and two types of pyrimidines, cytosine (C) and thymine (T). Thus, the above sequence might now be represented as (say) ACGATGCCGTA.... Chargaff's first parity rule is that purines pair with pyrimidines, specifically A with T and C with G. Thus, a duplex containing this sequence, with pairing between complementary bases in the "top" and "bottom" strands could be written as: |
| ACGATGCCGTAGCATCGT TGCTACGGCATCGTAGCA |
It was later realized that, under certain circumstances, this double helix could form "flaps". Thus each of the above two strands ("top" and "bottom") can form stem-loop secondary structures, of the following type, due to pairing with complementary bases in the same strand. |
|
C ACGATGC G TGCTACG T A |
For this to happen there have to be matching (complementary) bases. Only the bases in the loop (CGTA) are unpaired in this structure. The stem consists of paired bases. Thus Chargaff's parity rule has to apply, to a close approximation, to single strands of DNA. When one examines DNAs from whatever biological source, one invariably finds that the rule applies. We refer to this as Chargaff's second parity rule. Returning to our own written textual form of information, the sentence "Mary had a little lamb its fleece was white as snow" contains the information that a person called Mary is in possession of an immature sheep. The same information might be written in Chinese or Greek. Thus, the sentence contains not only its primary information, but also secondary information about its origin -- e.g. it is likely that the author is more familiar with English than other languages. Some believe that English is on the way to displacing other languages, so that eventually it (or the form it evolves to) will constitute the only language used by human beings on this planet. Similarly, in the course of early evolution possibly a prototypic nucleic acid language displaced contenders. It would be difficult to discern a relationship between the English, Chinese and Greek versions of the above sentence, because these languages diverged from primitive root languages thousands of years ago. However, in England, if a person with a Cockney accent were to speak the sentence it would sound like "Miree ader liawl laimb sfloyce wors woyt ers snaa". Cockney English and "regular" English diverged more recently and it is easy to discern similarities. Now look at the following text: |
| yewas
htbts llem ws arifea ac wMhitte alidsnoe la irsnwwis aee ar lal larfoMyce b sos woilmyt erdea |
One line of text is the regular English version with the letters shuffled. The other line is the cockney version with the letters shuffled. Can you tell which is which? If the shuffling was thorough, the primary information has been destroyed. However, there is still some information left. With the knowledge that cockneys tend to "drop" their Hs, it can be deduced that the upper text is more likely to be from someone who spoke regular English. With a longer text, this could be more precisely quantitated. Languages have characteristic letter frequencies. You can take a segment ("window") and count the various letters in that segment. In this way you can identify a text as English, Cockney, Chinese or Greek, without too much trouble. We can call this information "secondary information". There may be various other levels of information in a sequence of symbols. To evaluate the secondary information in DNA (with only four "letters"), you select a "window" (say 1000 bases) and counts the number of bases in that window. You can apply the same window to another section of the DNA, or to another DNA molecule from a different biological species, and repeat the count. Then you can compare DNA "accents". The best understood type of primary information in DNA is the information for proteins. The DNA sequence of bases (one type of "letter") encodes another type of "letter", the "amino acids". There are 20 amino acids, with names such as aspartate, glycine, phenylalanine, serine and valine (which are abbreviated as Asp, Gly, Phe, Ser and Val). Under instructions received from DNA, amino acids are joined together in the same order as they are encoded in DNA, to form proteins. The latter, chains of amino acids which fold in complicated ways, play a major role in determining how we interact with our environment. The proteins determine our "phenotype". For example, in an organism of a particular species ("A") the twenty one base DNA sequence: |
| TTTTCATTAGTTGGAGATAAA |
read in sets of three bases ("codons"), conveys primary information for a seven amino acid protein fragment (PheSerLeuValGlyAspLys). All members of the species will tend to have the same DNA sequence, and differences between members of the species will tend to be rare and of minor degree. If the protein is fundamental to cell function it is likely that organisms of another species ("B") will have DNA which encodes the same protein fragment. However, when we examine their DNA we might find major differences compared with the DNA of the first species (the similarities are emphasized in red): |
| TTCAGCCTCGTGGGGGACAAG |
This sequence also encodes the above protein fragment, showing that the DNA contains the same primary information as in the first DNA sequence, but it is "spoken" with a different "accent". This secondary information might have some biological role. It is theoretical possible (but unlikely) that all the genes in an organism of species B would have this "accent", yet otherwise encode the same proteins. In this case, organisms of species A and B would be both anatomically and functionally (physiologically) identical, while differing dramatically with respect to secondary information. On the other hand, consider a single change in the sequence of species A to: |
| TTTTCATTAGTTGGAGTTAAA |
|
Here the difference (emphasized in red) would change one of the seven amino acids. It is likely that such minor changes in a very small number of genes affecting development would be sufficient to cause anatomical and morphological differentiation within species A (e.g. compare a bulldog and a poodle, as "varieties" of dogs, which are able to breed with each other). Yet, in this case the secondary information would be hardly changed. The view developed in these pages is that, like the Cockney's dropped H's, the role of secondary information is to initiate, and, for a while, maintain, reproductive isolation. This can occur because the genetic code is a "redundant" or "degenerate" code; for example, the amino acid serine is not encoded by just one codon; there are six possible codons (TCT, TCC, TCA, TCG, AGT, AGC). In the first of the above DNA sequences (A) the amino acid serine (Ser) is encoded by TCA, whereas AGC is used in the second (B). On the other hand, the change in species A from GAT (first sequence) to GTT (third sequence) changes the encoded amino acid from aspartic acid (Asp) to valine (Val), and this should be sufficient to change the properties of the corresponding protein, and hence change the phenotype. Thus, the biological interest of linguistic barriers is that they also tend to be reproductive barriers. Even if a Chinese person and an English person are living in the same territory ("sympatrically"), if they do not speak the same language they are unlikely to marry. The Chinese tend to marry Chinese and produce more Chinese. The English tend to marry English and produce more English. Even in England, because of the "class" barriers so colourfully portrayed by George Bernard Shaw, Cockneys tend to marry Cockneys, and the essence of the barrier from people speaking "regular" English is the difference in accent. Because of other ("blending") factors at work in our society it is unlikely that this linguistic speciation will continue to the extent that Cockney will become an independent language. However, the point is that when there is "incipient" linguistic speciation, it is the secondary information (dropped H's), not the primary information, which constitutes the barrier. Before the genetic code was deciphered in the early 1960s, researchers such as Chargaff (1950), Wyatt (1952) and Sueoka (1961) studied the base composition of DNAs with a major interest in the primary information -- how a sequence of bases might be related to a sequence of amino acids. However, their results have turned out to be of greater interest with respect to the secondary information in DNA. That's all you need for an introduction. Enjoy! Donald Forsdyke |
 |
|
In 1950 Erwin Chargaff's enunciated his first parity rule for duplex DNA (A% = T% and G% = C%). In the 1960's came his second parity rule, namely that the first parity rule also applies, to a close approximation, to single-stranded DNA. Other rules are his cluster rule (that pyrimidines often occur in runs or clusters, and hence on the complementary strand purines do likewise) and his GC rule (that (G+C)% is relatively constant within a species, but often differs between species). Tongue in cheek, while dismissing "empty speculations," Chargaff (1950) claimed that he would not mention such fanciful terms as "template" - but in so doing, he succeeded in bringing the template idea to readers' attentions! And he concluded by noting that base composition was "a characteristic of the species, but not of the tissue, from which they [DNA samples] are derived." So DNA was possibly "the agent concerned with the transmission of inherited properties, [and] ... just as the deoxypentose nucleic acids [DNAs] of the nucleus are species-specific and concerned with the maintenance of the species, the pentose nucleic acids [RNAs] of the cytoplasm are organ-specific and involved in the important task of differentiation." |
|
In 1979 Chargaff came out of retirement and
discussed some of his work at a meeting on the history of molecular biology. His
colourful, characteristically poetical, address, entitled For a Summary of Chargaff's contributions, please see:
and the biographical sketch I wrote for
the |
|
Chargaff showed that base composition, expressed as (G+C)%, tends to be constant within a species (i.e. it is a species characteristic).
In 1961, Sueoka (like Wyatt in 1952) was interested in how DNA encodes its primary information (e.g. protein sequence). He dismissed (G+C)% as essentially a random (stochastic) phenomenon, of no fundamental importance (see "On the genetic basis of variation and heterogeneity of DNA base composition".1962; Proc. Natl. Acad. Sci. 48, 582-592). He maintained this view until at least 1995. However, even in 1961 (Cold Spring Harbor Symposium 26, 35-43) for strains of Tetrahymena Sueoka noted a link of "GC" [= (G+C)%)] with reproductive isolation: |
| "DNA base composition is a reflection of phylogenetic relationship. Furthermore, it is evident that those strains which mate with one another (i.e. strains within the same 'variety') have similar base compositions. Thus strains of variety 1 ..., which are freely intercrossed have similar mean GC content. ... If one compares the distribution of DNA molecules of Tetrahymena strains of different mean GC contents, it is clear that the difference in mean values is due to a rather uniform difference of GC content in individual molecules. In other words, assuming that strains of Tetrahymena have a common phylogenetic origin, when the GC content of DNA of a particular strain changes, all the molecules undergo increases or decreases of GC pairs in similar amounts. This result is consistent with the idea that the base composition is rather uniform not only among DNA molecules of an organism, but also with respect to different parts of a given molecule." |
|
In these pages I point to (G+C)% as a "second level" of genetic information of great relevance to the question of the origin of species (Forsdyke, 1996a). Whatever the truth of the matter, an important 1961 paper of Sueoka (below), clearly demonstrated the power of apparently internal genomic forces over external forces (e.g. conventional natural selection), in the moulding of genomes. As usual, the story can be traced back to 19th century origins. Of Darwin's proposal as to how species originate, Mendel is reported (Iltis 1932) to have said: |
|
"This much already seems clear to me, that nature [natural selection] does not modify species in any such way, so some other force must be at work". |
|
In his letters to Nageli, a botany professor in Munich, Mendel elaborated on this "force": |
"If the change of environment of growth [phenotypic selective factors] were the sole cause of variability, we should expect that those domesticated plants which had been cultivated for many centuries under almost identical conditions would by now have recovered their constancy [stopped varying]. But this, as we know, is not the case, for it is among these plants that we find, not only the most diversified, but also the most variable forms. ... It seems more probable that, in this matter of the variability of domesticated plants, there has been at work a factor to which hitherto little attention has been paid." |
|
Agreeing with Mendel, Nageli is reported (Iltis 1932) to have: |
"... rejected ... [natural] selection as an active factor in the origin of species, thus setting himself in strong opposition to Darwin. In his view the progressive change in living forms was to be explained as the expression of a progressive tendency inherent in living matter, a tendency which was not to be regarded mystically, but as dependent upon mechanical [chemical?] forces". |
Thus, Nageli took Mendel's criticism one step further in suggesting (vague though it might be) an internal "mechanical" cause of the origin of species, independent of natural selection. A 20th century clue was provided by Sueoka's demonstration (1961) that the composition of proteins is vulnerable to such internal forces. A protein has a job to do. Its sequence, and hence structure, should subserve this role. Under the struggle for existence, an organism with a more efficient protein should win out. Thus, there should have been a high evolutionary premium in modifying a protein to maximize efficiency. If a protein worked marginally better with the basic amino acid arginine than with the basic amino acid lysine in a certain position, then one might imagine that evolutionary selection pressures would favour organisms in which the position contained arginine. However, lysine is encoded by GC-poor codons (AAA, AAG), whereas arginine can be encoded by GC-rich codons. Thus, by switching between amino acids an organism would be able to respond to internal (intrinsic) genomic forces, while perhaps not greatly decreasing its ability to adapt to conventional Darwinian (extrinsic) environmental forces. Sueoka's work, produced shortly before the genetic code was deciphered, showed how this might have occurred. Of course, the degeneracy of the genetic code would have allowed the composition of DNA to respond to hypothetical internal genomic forces without necessarily changing amino acid composition ("synonymous" mutations). But if this flexibility were insufficient, amino acids themselves might have to change ("non-synonymous" mutations). Evidence for this change would indirectly provide evidence for the existence of such apparently internal genomic forces. Thus instead of the simple evolutionary chain of command: |
| ENVIRONMENT --> protein --> DNA |
| there was also the possibility of the reverse evolutionary chain of command: |
| DNA --> protein --> ENVIRONMENT |
|
Later work (discussed below) showed that the structures of certain RNAs also appeared to have responded to intrinsic, rather than conventional environmental forces. Thus, Ball (1973) concluded: |
"The amino acid sequence of MS2 [a virus whose genome is RNA]... has been subjected, during its evolution, to rearrangement in the interests of the secondary structure of the messenger RNA. This indicates that there is a pressure for some amino acid sequences to be selected according to criteria which are distinct from the structure and function of the protein they constitute". |
|
Finally Richard Grantham wrapped it all up with his "genome hypothesis", (Click Here) noting (1986): |
"According to the genome hypothesis each ... species has a 'system' or coding strategy for choosing among synonymous codons. This system or dialect is repeated in each gene of a genome and hence is a characteristic of the genome." |
|
Through the work of Akiyoshi Wada and Georgio Bernardi and their colleagues, we now better appreciate that, although this is true, individual genes ("microisochores") or genome sectors ("macroisochores") have their own sub-dialects, which vary around the average for the species. Thus dialects can distinguish both species within a phylogenetic group, and genes within a species. The following figure summarizes some of the multiple pressures, acting just at the level of messenger RNA (red line), which are described on these web-pages:
|
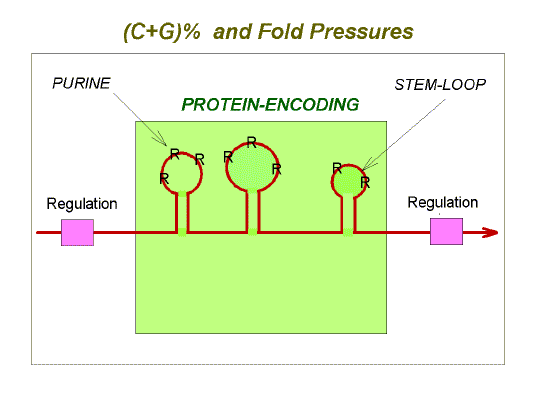 |
Note the repeated use of the phrases "conventional environment" and "conventional Darwinian natural selection". The conventional (i.e. "classical") environment acts on the conventional phenotype resulting in conventional natural selection/isolation. However, there is also a reproductive environment, namely the potential mates with which an organism may pair and reproduce its kind. Within a species there is the possibility of divergence (into dialects) at the genomic level, a process with consequences most manifest when meiosis occurs in the gonads of sons and daughters. This can result in a degree of infertility (hybid sterility) in the offspring of a cross between disparate types (whose dialects vary slightly from each other). Thus, it is appropriate to speak of a "genome phenotype". The reproductive environment (the genome phenotype of another) acts on an organism's own genome phenotype resulting in reproductive (physiological) selection/isolation. The resulting changes in DNA can, to a degree, influence protein function and hence the way the organism interacts with the conventional environment. Thus the reproductive environment, through ones "reprotype", impacts the conventional environment through one's "phenotype". |
|
Reproductive Environment ---> DNA ---> Protein ---> Conventional Environment |
|
Giorgio Bernardi and Giacomo Bernardi had some sense of this (1986; J. Mol. Evol. 24, 1-11) noting: |
"The organismal phenotype comprises two components, the classical phenotype, corresponding to the 'gene products', and a 'genome phenotype', which is defined by the [base] compositional constraints." |
For more on reproduction and "molecular sex" Click Here.
|
Correlation between Base Composition of Deoxyribonucleic Acid and Amino Acid Composition of Protein Proceedings of the National Academy of Sciences, USA (1961) 47, 1141-1149. By NOBORU SUEOKA DEPARTMENT OF MICROBIOLOGY, UNIVERSITY OF ILLINOIS Communicated by A. D. Hershey, June 1, 1961
by A. D. Hershey, June 1, 1961 [With copyright permission from Noboru Sueoka.] |
|
|
It has been known that the base composition of deoxyribonucleic acid (DNA) is quite variable from organism to organism. A good example of this variability is provided by bacterial species. Here, the mean guanine-cytosine content ranges from 26 to 74 per cent (1, 2). Moreover, the heterogeneity of base composition of DNA molecules from each species is relatively small (3-6). These observations would appear to provide a fruitful basis for comparative biological investigations in molecular terms. In the current views of DNA-protein coding, it is assumed that the base sequence of DNA has a direct correspondence with amino acid sequence in protein. However, whether the implied correlations between DNA base composition and amino acid composition of protein exist or not depends upon the mechanism of coding, its universality, and the magnitudes of the participating fractions of DNA and protein in the coding relation [Not known at the time of writing.]. If the majority of proteins do have coding relations with DNA, and if, in particular, the code is universal and four-symboled, we may expect some correlations between DNA and protein in their composition [Thus, the declared goal of Sueoka's work was directed towards the then very important coding problem (the primary information problem), rather than general aspects of evolution (the secondary information problem.]. From these points of view, a search for possible correlations between DNA base composition and amino acid composition of protein has been initiated. In the present paper, the results of the first stage of our work which is concerned with total protein (microsomal plus soluble) of bacterial cells will be presented. Amino acid compositions of bulk protein preparations isolated from 11 bacterial species whose DNA base composition varies from 35 to 72 per cent in guanine-cytosine content have been examined. The results indicate that correlations between component compositions of DNA and protein do indeed exist. |
|
Materials and Methods.- Most bacterial species used in the present investigation have been obtained from the American Type Culture Collection. One liter of an enriched nutrient broth medium (7) was inoculated and incubated at 37oC for about 15 hr with constant shaking. The minimal medium used was M 26 basal minimal medium (8). This medium was supplemented with glutamic acid (0.5 gm per liter) for Bacillus subtilis and Serratia marcescens. Cells were concentrated by centrifugation at 4,000 g for 15 min at 5oC, washed once with tris magnesium buffer (10-2 M Tris (hydroxymethyl) aminomethane plus 10-3 M Mg, pH 7.3), and suspended in the same buffer to make the final volume 20 ml. In order to disrupt the cells, the cell suspension was put through a French press twice under a pressure of 12,000 lb/inch2, keeping the system as cold as possible. The resulting suspension was centrifuged at 6000 g for 15 min at 5o C, and the supernatant (a crude extract) was saved. "Preparation 1" was obtained by heating the crude extract at 100o C for 10 min and then by dialyzing against three liters of distilled water for 24 hr at 4o C with constant stirring. The external water was changed once. A further purification of the crude extract for "preparation 2" was achieved by adding urea to a final concentration of 4.5 M, and by stirring the solution at room temperature for 30 min. Ammonium sulfate (0.6 gm per ml) was added to the mixture, and the solution was centrifuged at 12,000 g for 20 min at 5o C. The precipitate was washed with 20 ml of 90% ethanol twice, suspended in 10 ml of distilled water, and dialyzed against 3 liters of distilled water for 24 hr at 4o C with constant stirring, The external water was changed once. The resulting preparation is called "preparation 2." The amounts of protein of preparations 1 and 2 were determined by the Biuret method. The contents of DNA and RNA were determined by indole(9) and orcinol (10) methods, respectively. These are expressed as weight per cent of protein (Tables 1 and 2). On the average, 2.8 per cent of DNA and 16 percent of RNA were found in the preparations listed in Table l. For aminoacid analyses, the procedure of reference 11 was followed. An aliquot of preparation 2 containing 5 mg of protein was taken in a test tube, and water was added to make the volume 2 ml. Two milliliters of concentrated HCl (12 N) was added to it, and the tube was sealed under vacuum after the sample was frozen. The sealed tube was kept in an air-drying oven at 110 oC for 22 hr. The hydrolyzate was dried and redissolved in 5 ml of a 0.2 N sodium citrate buffer, pH 2.2(11). Minute amounts of carbon appeared after hydrolysis. This was removed by filtration. The amino acid composition was analyzed by a SPINCO Automatic Amino Acid analyzer. In all but three of the samples analyzed, unknown ninhydrin positive peaks were quite rare, and they occupied less than 1 per cent of the total peak area. The exceptions were Serratia marcescens grown on either enriched broth medium (1.1%) or minimal medium (1.1%), and one culture of Micrococcus lysodeikticus grown on enriched broth medium (l.0%). |
|
Results - The relative molar content of each amino acid was expressed in the following manner: molar amounts of amino acids, lysine, histidine, arginine, aspartic acid and asparagine, glutamic acid and glutamine, proline, alanine, valine, leucine, tyrosine, and phenylalanine, which are known to be stable and to be well recoverable in the analysis (12), were summed. Other amino acids including glycine, threonine, serine, isoleucine, methionine, and cysteine are classified as "unstable amino acids." Glycine was not included in the "stable" class, because some glycine is produced by the decomposition of contaminating nucleic acids (13). It is evident from the data of Tables 1 and 2 [not shown here] that no correlations exist between the extent of contamination with nucleic acid and the observed amino acid composition of the different protein preparations. Subsequently, the amount of each amino acid, both "stable" and "unstable," was expressed by its proportion to the sum of the stable amino acids (14). The values expressed in this way are presented in Table 1 and Figure 1. |
|
|
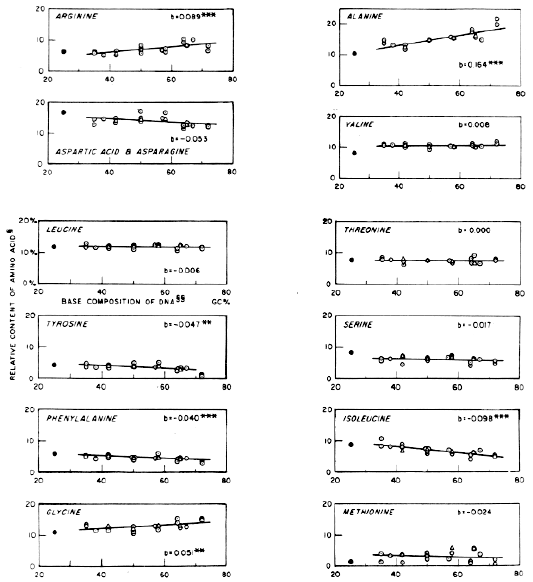
FIG. 1.- Relation between DNA base composition and amino acid composition of protein in bacteria. The relative molar content of each amino acid is expressed as a percentage of the amino acid content over the sum of stable amino acids: lysine, histidine, arginine, aspartic acid plus asparagine, glutamic acid plus glutamine, proline, alanine, valine, leucine, tyrosine, and phenylalanine. The base composition of DNA is expressed as a molar content (%) of guanine plus cytosine (GC). The GC contents of bacterial species adopted in this work are the data of reference 1. Regression coefficient (b). Tetrahymena data are not included in the calculation. Levels of significance of b's (or slope) are shown by asterisks (one asterisk = significant 0.02<P<0.05; two asterisks= highly significant 0.001<P<0.02; three asterisks= extremely significant P<0.001). |
|
The base compositions of DNA of bacteria used are taken from reference 1 and expressed as molar guanine-cytosine content in percentage. The above way of calculation has several advantages which may be listed as follows. (a) It obviates the error in estimating the relative amounts of the stable amino acids stemming from poor recoveries of unstable amino acids. (b) Relative contents of all amino acids are not changed after the calculation. (c) The sum of the "stable" amino acids is used as the denominator, and these have both positive and negative regression coefficients. Moreover, "stable" amino acids, the sum of which is used as a common denominator, include a majority (thirteen) of the twenty amino acids. As a consequence, the calculation will hardly affect the regression coefficients for the unstable amino acids. Although there is no special reason to choose linear relations between the GC content of DNA and the amino acid content of protein, a straight line was simulated by least square methods. A summary of the statistical analysis is given in Table 3. Several amino acids show significant regression coefficients, both positive and negative. |
| Amino acid | Average content (yaver) | Regression coefficient (b) | Confidence limits of b (5% level) | Probability (P) | Extrapolation of GC content to: | |
| 0% | 100% | |||||
| Stable | ||||||
| Lys | 8.46 | -0.084 | 0.046 | 0.01-0.001 | 12.96 | 4.56 |
| His | 2.48 | -0.010 | 0.015 | 0.10-0.20 | 3.38 | 2.38 |
| Arg | 7.23 | +0.089 | 0.043 | <0.001 | 2.46 | 11.36 |
| AspX | 13.85 | -0.053 | 0.054 | 0.10-0.05 | 16.69 | 11.39 |
| GluX | 15.77 | -0.052 | 0.050 | 0.05-0.10 | 18.14 | 12.98 |
| Pro | 5.89 | +0.024 | 0.020 | 0.02 | 4.60 | 7.00 |
| Ala | 15.30 | +0.164 | 0.055 | <0.001 | 6.51 | 22.91 |
| Val | 10.39 | +0.008 | 0.023 | 0.30-0.50 | 9.96 | 10.76 |
| Leu | 11.90 | -0.006 | 0.020 | 0.50-0.70 | 12.22 | 11.62 |
| Tyr | 3.63 | -0.047 | 0.035 | 0.01-0.02 | 6.15 | 1.45 |
| Phe | 4.68 | -0.040 | 0.022 | <0.001 | 6.82 | 2.82 |
| Unstable | ||||||
| Gly | 12.80 | +0.051 | 0.039 | 0.01-0.02 | 10.07 | 15.17 |
| Thr | 7.61 | +0.0001 | 0.027 | 0.90 | 7.60 | 7.62 |
| Ser | 6.11 | -0.017 | 0.033 | 0.30-0.50 | 7.12 | 5.32 |
| Met | 3.07 | -0.024 | 0.062 | 0.30-0.50 | 4.36 | 1.96 |
| Ileu | 7.06 | -0.098 | 0.032 | <0.001 | 12.31 | 2.51 |
Least square method was used to calculate a and b for a linear relation y = a + b(x-xaver). Here y is a relative molar content of an amino acid, and yaver, its average, while x and xaver are GC content of DNA and its average. It is noted that a is equal to yaver, the average of y. The way of regression analysis used here is described in reference 21. In calculating regression coefficients all but the Tetrahymena data were included. |
||||||
|
The relative amino acid content is quite reproducible, which is indicated by the results on duplicate cultures of most species. The effect of environmental differences on the relative amino acid content of the total protein was examined by changing the composition of medium and the culture temperature and by comparing logarithmic and stationary phases of growth (Tables I and 2 [Not shown]). No significant differences were observed. The present data on the amino acid composition of E. coli protein agree fairly well with those obtained by Roberts et al.(15; Table 2). Selective enrichment of particular amino acids during the protein isolation procedure seems to be unlikely from the data on preparations 1 and 2 in E.coli. The relative amounts of both fractions are very similar. |
|
Discussion - Several interesting features emerge from this investigation.
The correlation disclosed here may not, however, represent the true picture for coded protein, since some noncoded proteins may exist. Consequently, the correlations reported here may represent underestimations of the true values. When more data on amino acid composition of different cellular components become available, we will be in a better position to evaluate this evidence in relation to the coding problem. For this reason, any detailed deductions of a coding model from the present data are not attempted in this paper. Nevertheless, some qualitative discussions will be attempted with the hope that they may stimulate further investigations along this line. (1) The present data seem to support universality of the code among bacteria. The presence of different codes among the bacteria would clearly preclude finding any correlation. Relevant to this issue are the preliminary data on amino acid content of total protein of Tetrahymena pyriformis included in Table 1. T. pyriformis strain 1A has 25 per cent GC content with a small heterogeneity (16). The amino acid content of its protein fits remarkably well with the extrapolated lines of the data obtained with the bacteria (see Table 1). This may suggest that the underlying coding is also common to protozoa. (2) The argument that the wide variation of DNA base composition is due to the presence of large amounts of "nonsense" DNA (4, 17) seems to be rather unlikely because of the presence of correlations. This is particularly true of the correlations observed with arginine, tyrosine, and isoleucine, where the extrapolation to 0 or 100 per cent GC content makes the amino acid content almost zero. (3) The "6 keto-6 amino two-symbol code" (4, 18) is not supported by the present data, for in such a system we would expect no correlations. (4) If the coding ratio is universal and relatively small (e. g., 3, 4, 5, 6, etc.), it seems logical to have even numbers, namely 4 or 6, rather than odd numbers to explain the existence of amino acids which show neither positive nor negative correlations. To illustrate this point, let us take triplet and quadruplet nonoverlapping four-symbol codes (19, 20). Since our analysis is based on the GC content of DNA, two base pairs, A-T and T-A, are indistinguishable, and they are expressed as a-pairs. Similarly G-C and C-G pairs are denoted as g-pairs. Following this terminology, we can classify the possible triplets into two classes, exclusive and asymmetric. The triplets of the exclusive class are those which contain only a or g , and we may designate them by the symbols, a3 and g3. Analogously, asymmetric triplets are ones which have both a and g but in different numbers, namely a2g and ag2. Following a similar argument, we can classify the quadruplets into three classes, exclusive(a4 and g4) , asymmetric (a3g and ag3), and symmetric (a2g 2) The symmetric class consists of quadruplets with equal numbers of a 's and g 's. Then the frequency of each type of triplet or quadruplet will have correlations of different signs and magnitudes with the GC content of DNA depending on the content of g in the particular type. Accordingly, the same kinds of correlations will be expected between amino acids in protein corresponding to each triplet (or quadruplet) and the GC content of DNA (Table 4 [Not shown]). It is noted that the symmetric class will have no correlation when a linear regression line is estimated. The expected correlations within our GC range of analysis (25 to 75 per cent) are shown in Table 4. If we force our data to this scheme, the best fit is given by assignments similar to that in Table 4. Needless to say, the validity of these issues must be subjected to further experimental tests. Nevertheless, the results are encouraging for our faith in the existence of a coding mechanism amenable to an ultimate resolution. |
Summary - Amino acid composition of the bulk protein isolated from 11 bacterial species whose DNA base compositions vary from 35 to 72 per cent in guanine-cytosine content has been examined. The following points are of interest.
The results obtained were discussed in relation to the coding problem. |
|
I wish to thank Mrs. C. W. Luker and Mr. R. Igarashi for their help with the amino acid analysis and Mrs. M. E. McCarty for her aid in preparation of the manuscript. I am grateful to Drs. S. Spiegelman and A. D. Hershey for reading and criticizing the manuscript. This work was supported by a grant from the National Science Foundation (G-15080).
|
Conflict Between Coding Potential and Secondary Structure
The above work of Sueoka (1961) first suggested that the amino acid sequence of a protein (and hence potentially the properties of that protein), responds not only to the forces of conventional Darwinian natural selection, but also the intragenomic force "(C+G)% pressure" or "GC pressure". Indeed, when the first partial sequences of genomes became available a decade later, a force possibly involved in recombination was identified, -- "fold pressure", or "stem-loop" pressure. In 1968 Robin Holliday drew attention to the possibility that genomic sequences referred to as "recombinators" might overlap sequences concerned with other functions, such as protein synthesis :
Among the first genomes to be sequenced were those of the single stranded RNA phages [phages whose genomes are RNA-based], such as R17. It was noted that there was far more potential secondary structure than expected on a random basis. In the discussion at the Cold Spring Harbor Laboratory Symposium after presentation of sequences from R17 (Jeppesen et al. 1970), Winston Salser noted: |
"As shown by the preceding paper, RNA phage R17 has very extensive
regions of highly ordered base pairing. It has seemed likely that this might be necessary
to allow phage packaging. Bernice Ricard and I were therefore somewhat surprised to find
that [DNA phage] T4
messengers [mRNAs
], which do not have to be
packaged, also have a very large amount of secondary structure. For
instance, the ratio of sedimentation velocity in normal sucrose gradients to sedimentation
velocity in 99% dimethyl sulfoxide gradients (completely denaturing conditions) is the
same for the active lysozyme messenger and for 16 S ribosomal RNA (rRNA), which also has
extensive base-pairing (Fellner et al., this volume). [In
sucrose secondary structure is maintained, whereas in dimethyl sulfoxide the molecule is
'denatured', meaning secondary structure is lost.]
Bernice Ricard has also investigated the hypochromicity of bulk T4 messenger RNA (mRNA)
purified free from all E. coli stable RNA species by chromatography ... . The
sized messenger fractions all have hypochromicities ... at least 80% as great as those
observed for 16 S rRNA. [When nucleic acid goes from
single-strand form to duplex form there is a loss of optical extinction at 255 mu,
referred to as hypochromicity, which can be used to quantitate secondary structure.] Even more important, the Tm's [Temperatures at which nucleic acid duplexes "melt" into single stranded forms] of the mRNA fractions are within two degrees of those for 16 S RNA. Since these mRNAs, unlike R17, do not have to be packaged into phage particles, our results suggest that a high degree of secondary structure may be important in the functioning of most mRNA molecules. Because of the very high Tm's we do not think that the base pairing seen is random. The possible functions of such extensive regions of base-pairing are unknown." |
Initially this was interpreted as a phenomenon primarily operative at the mRNA level, rather than at the genome level. Crick (1971) in his "unpairing hypothesis" proposed that, without strand breakage, duplex DNA might open up to expose single-stranded segments which would be available for pairing with, and hence recognition of, a homologous segment borne by a homologous chromosome. Sobell (1972) and others, pointed out that single-stranded segments would adopt temporary stem-loop configurations, implying that Holliday's recombinator sequences might merely be sequences adapted to form such structures. In that these structures would facilitate the homology search necessary for homologous recombination, there would be a selective pressure for their retention, perhaps at the expense of co-encoded protein sequences if the recombinators happened to be intragenic. Although sharing with Holliday the belief that recombinator sequences were primarily adaptations for recognition by proteins, rather than for recognition by homologous DNA, Tamar Schaap in a paper entitled "Dual Information in DNA and the Evolution of the Genetic Code" (J. Theor. Biol. 32, 293-298; Click Here), pointed out that a mutation in a region of DNA concerned with recombination might both change an amino acid and affect the local frequency of recombination; this was a powerful counterargument to the view (Kimura and others) that mutations not affecting amino acid sequences are, by default, "neutral:"
|
|
As touched on above, this was further explored by L. Andrew Ball, who concluded (1972; J. Theor. Biol. 36, 313-320): |
"The physical properties and nucleotide sequences of some messenger RNA molecules suggest the presence of extensive base-paired secondary structure. This paper evaluates the evolutionary conflict between this secondary structure and the encoded amino acid sequence, and concludes that selection pressure for specific base pairing in a messenger RNA severely limits its coding potential. |
 |
|
Elsewhere he noted concerning phage MS2 (1972; Nature New Biology 242, 44-45): |
"Because the base-pairing is too extensive overall to be accidental, elimination of [coding] degeneracy as an exceptional quantitative contributor to optimization [of secondary structure] implies that selection pressure has optimized pairing between non-degenerate positions. In other words, the amino acid sequence of MS2 coat protein has been subjected, during its evolution, to rearrangement in the interests of the secondary structure of the messenger RNA. This indicates that there is pressure for some amino acid sequences to be selected according to criteria which are distinct from the structure and function of the protein they constitute." |
|
From further bioinformatic analysis of MS2 he concluded (1973; J. Theor. Biol. 41, 243-247): |
"The presence of secondary structure in a messenger RNA suggests that natural selection has moulded the nucleotide sequence to meet the demands of base-pairing as well as those of the encoded amino acid sequence. Despite the degeneracy of the genetic code, neither requirement can be fulfilled independently of the other, so that evolved RNA and protein structures must represent a compromise. One feature of this compromise is illustrated by the coat protein of bacteriophage MS2. Those amino acid residues which, when represented by base-paired codons, impose the most severe limitations on the overall protein sequence, show a clear tendency to avoid being encoded in base-paired regions of the messenger RNA." |
This has been supported by subsequent studies, but the implication that "the interests of the secondary structure of the messenger RNA" is the main driving force has been challenged. As indicated above, although most mRNAs show much more secondary structure than expected on a random basis, this may reflect pressures operating at the level of the DNAs from which the RNAs are transcribed. As documented in these web-pages, studies from the laboratories of J. V. Maizel and of the present author, show that highly significant stem-loop potential is widely distributed in genomes, both in protein-coding regions and in non-protein-coding regions (intergenic DNA and introns). This partly explains the generality of Chargaff's second parity rule. Indeed, looking at both phage and human RNA sequences White et al. noted in 1972 (Science 175, 1264):
|
In view of the adaptive pressures from extrinsic (environmental) and intrinsic (internal, and particularly, genomic) forces, it is hard to imagine any mutation as really "neutral" (see Codon Choice and Grantham's "genome hypothesis"; (Click Here)). Indeed, from the first it was recognized that the rate and/or accuracy of translation of a mRNA was partly determined by which of a choice of codons (between one to six) was employed to encode a particular amino acid. In microorganisms a high rate of protein synthesis can be evolutionarily advantageous, so microorganisms tend to prefer codons that can be translated most rapidly. Sometimes, a slowing of the rate of protein synthesis is advantageous - in allowing the correct folding of a protein - so in this circumstance there will be a selection pressure to prefer codons that can be translated most slowly. However, the "neutralists" long hold the high ground, as the following exchange relates. |
![]()
Neutralism Versus Selectionism:
Genomic G+C content, RNA secondary structures, and optimal growth temperature in prokaryotes
In a paper in the Journal of Molecular Evolution in 1997, a viewpoint contained in these web pages was criticized by Galtier and Lobry. On the advice of his reviewers, the following rebuttal (a copy of which was sent to Dr. Lobry), was declined by Professor Bernardi for publication in the Journal of Molecular Evolution as a LETTER TO THE EDITOR. It was published by the author on the Bionet.molbio.evolution web-page on 26th February 1999, and here in March 1999. Professor Bernardi (1999, 2000) supported the arguments presented here (see below). |
| Professor Giorgio
Bernardi, Associate Editor, Journal of Molecular Evolution Laboratoire de Genetique Moleculaire, Institut Jaques Monod, 2 Place Jussieu, 75005 Paris, France [Receipt acknowledged July 15th 1997] Letter to the Editor concerning paper: Relationships between genomic G+C content, RNA secondary structures, and optimal growth temperature in prokaryotes, by N. Gautier & J. R. Lobry (1997) J. Mol. Evol. 44, 632-636. Dear Professor Bernardi,The above paper is held by the authors to support the neutral explanation (proposed by Lobry), rather than the selectionist explanation (proposed by me), for why Chargaff's parity rule applies to single stranded DNA. It is presumably for this reason that you asked me to review the paper for JME last fall. I recommended that the paper be accepted because it confirmed previous studies on a wider range of species. However, I had reservations about the authors' logic with respect to whether their work supported neutralist or selectionist viewpoints, and made suggestions for revisions in this respect. I note from the paper which has recently appeared, that the suggestions have not been adopted, and so the paper still contains major flaws, which might mislead a less diligent reader. Accordingly, I have written a short letter which I believe offers a better interpretation of the data of Gautier and Lobry. Please will it be considered for publication in JME. Yours sincerely, Donald R. Forsdyke Enclosed: 3 copies of paper: "Neutralism versus selectionism: no correlation between optimum growth temperature and genome G+C content does not refute the selectionist viewpoint". |
Neutralism versus selectionism: no correlation between optimum growth temperature and genome G+C content does not refute the selectionist viewpoint
Donald R. Forsdyke
Department of Biochemistry, Queen's University, Kingston, Ontario, Canada K7L3N6
Abstract Galtier and Lobry (1997; J Mol Evol 44:632-636) compared the optimum growth temperatures of various prokaryotes with the G+C content of their genomic DNA and of various non-mRNA RNA species (e.g. ribosomal RNAs). Since GC bonds confer greater stability on nucleic acid secondary structure than AT bonds, their data strongly suggest that an increase of G+C content is needed for the stabilization at high temperature of rRNA secondary structure (stem-loops), but not of DNA secondary structures. The authors propose that "any secondary structure that must endure at high temperatures requires a high G+C content", so that "a high proportion" of stem-loop "secondary structures in bacterial genomes is unlikely". Thus, the fact that Chargaff's parity rule (%A=%T, %G=%C) applies to single-stranded DNA (as to single-stranded RNA), is held to be "poorly explained" on the basis of an evolutionary pressure on DNA to form stem-loops (as proposed by Forsdyke 1995; J Mol Evol 41:573-581). Rather the parity rule would be explained by "neutral directional mutational pressure" (Lobry, 1995; J Mol Evol 40:326-330). Key words: Chargaff's parity rule - G+C content - Secondary structure |
A single-stranded nucleic acid with the potential to form stem-loop secondary structures should tend to follow Chargaff's parity rule (%A=%T, %G=%C). This is because Watson-Crick base pairing is involved in the formation of stems, and might also be involved in intrastrand loop-loop interactions which would further stabilize the secondary structures. The potential for genomic DNA in the classical duplex form to extrude stem-loop secondary structures is widely distributed in the DNA of all species studied (Murchie et al. 1992; Forsdyke, 1995a), and involves both extragenic DNA, and intragenic DNA (Forsdyke, 1995b,c; 1996a,b). Thus, the compositions of single-stranded DNAs tend to follow the parity rule (Forsdyke, 1995c), and the RNAs transcribed from them might also tend to follow the rule. Indeed, mRNAs, which in prokaryotes with compact genomes are more likely to be reflective of total genomic structure and base composition than rRNAs (Muto and Osawa 1987), can be folded using energy-minimization algorithms into very compact stem-loop secondary structures (Jaeger et al. 1990). Ribosomal RNAs also form compact stem-loop secondary structures and so tend to follow Chargaff's parity rule. In prokaryotes optimal growth temperature (Topt) correlates positively with the G+C content of rRNA, but not of genomic DNA (Dalgaard and Garret 1993). This has been confirmed by Galtier and Lobry (1997), who propose, quite plausibly, that the increase in G+C content of rRNA is required to stabilize stem-loop structures under hyperthermic conditions. Free of coding constraints, yet required to form part of the presumably very precise structure of ribosomes, rRNA might be under greater pressure to accept mutations which increase G+C content than most mRNAs. However Galtier and Lobry (1997) also propose that "any secondary structure that must endure a high temperature requires a high G+C content" (my italics). This is manifestly incorrect, since the authors themselves report that "no correlation was found between genomic G+C content and Topt". It follows that the classical secondary structure of duplex DNA is likely to be stabilized by what the authors refer to as "other physiological adaptations"; these might include increased association of DNA with basic polyamines (Oshima et al. 1990), and relaxation of supercoiling (Friedman et al. 1995). In the context of possible DNA stem-loop secondary structures, Galtier and Lobry (1997) conclude that "a high proportion of [such] secondary structures in bacterial genomes is unlikely" so that the fact that Chargaff's parity rule applies to single-strands of DNA "is poorly explained" (by Forsdyke 1995c) on this basis. Instead, the authors propose that Chargaff's parity rule tends to apply to single-stranded DNA because of neutral directional mutation pressure (Lobry, 1995; Sueoka, 1995). The arguments of Galtier and Lobry (1997) rest on the assumption that stem-loop structures in rRNAs are comparable with stem-loop structures extruded from duplex DNA. However, there is no reason to believe that "other physiological adaptations" at high temperatures cannot stabilize both the classical DNA duplex secondary structure and DNA stem-loop secondary structures. The energetics of helix (stem) formation is essentially the same whether DNA has a classical duplex secondary structure or a stem-loop secondary structure (Murchie et al. 1992). As we currently understand it, the latter structure would be required only under certain clearly defined, but selectively important, circumstances (i. e. for recombination repair; Forsdyke, 1996b; Kleckner, 1997). The enduring DNA secondary structure would be the classical duplex form. Thus, Galtier and Lobry (1997) do not make an adequate case for neutral directional mutational pressure as an explanation for the applicability of Chargaff parity rule to single-stranded DNA. In the context of their data, selectionist arguments are no less probable than neutralist arguments. The data indicating no selective advantage of high genomic G+C content at high temperature (Fiala and Stetter 1986; Galtier and Lobry 1997), appear to support the neutralist argument that variations in genomic G+C content have arisen by drift in small populations (Filipski 1990). However, the data are also consistent with the selectionist argument that genomic G+C content is too important merely to follow the dictates of temperature, since its primary role is to respond to the G+C contents of other species in order to prevent inter-species recombination (Forsdyke, 1996b). Other selectionist arguments have been advanced by Bernardi (1993). References Bernardi G (1993) The vertebrate genome. Isochores and evolution. Mol Biol Evol 10:186-204 Dalgaard JZ, Garrett A (1993) Archaeal hyperthermophile genes. In: Kates M et al. (eds) The biochemistry of Archaea (Archaebacteria). Elsevier Science, Amsterdam, pp 535-562 Filipski J (1990) Evolution of DNA sequences. Contributions of mutational bias and selection to the origin of chromosomal compartments. Adv Mut Res 2:1-54 Forsdyke DR (1995a) A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol Biol Evol 12:949-958 Forsdyke DR (1995b) Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. An application of FORS-D analysis. Mol Biol Evol 12: 1157-1165 Forsdyke DR (1995c) Relative roles of primary sequence and (G+C)% in determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J Mol Evol 41:573-581 Forsdyke DR (1996a) Stem-loop potential in MHC genes: a new way of evaluating positive Darwinian selection. Immunogenetics 43:182-189 Forsdyke DR (1996b) Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths". J Theor Biol 178:405-417 Friedman SM, Malik M, Drlica K (1995) DNA supercoiling in a thermotolerant mutant of Escherichia coli. Mol Gen Genet 248:417-422 Galtier N, Lobry JR (1997) Relationships between genomic G+C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J Mol Evol 44:632-636 Jaeger JA, Turner DH, Zuker M (1990) Predicting optimal and suboptimal secondary structure for RNA. Meth Enzymol 183:281-317 Kleckner N (1997) Interactions between and along chromosomes during meiosis. Harvey Lectures 91:21-45 Lobry JR (1995) Properties of a general model of DNA evolution under no-strand-bias conditions. J Mol Evol 40:326-330 Murchie AIH, Bowater R, Aboul-Ela F, Lilley DMJ (1992) Helix opening transitions in supercoiled DNA. Biochem. Biophys. Acta 1131:1-15 Muto A, Osawa S (1987) The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci USA 84:166-169 Oshima T, Hamasaki N, Uzawa T, Friedman SM (1990) Biochemical functions of unusual polyamines found in the cells of extreme thermophiles. In: Goldembeg SH, Algranati ID (eds) The biology and chemistry of polyamines. Oxford University Press, New York, pp 1-10 Sueoka N (1995) Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J Mol Evol 40:318-325 The above arguments concerning the Galtier-Lobry paper were in accord with Professor Bernardi's views (2000; Gene 241, 3-17):
Essentially the same statement had been made
by Bernardi in 1993 (Molecular Biology &
Evolution 10, 186-204; see p. 201 "In
addition, thermal stabilization of genomes might For further discussion of Thermophiles and DNA structure see "Chargaff's Legacy" (2001) (Click Here), and Lambros et al. (2003) (Click Here). Note also "High Affinity DNA Binding of HU Protein from the Hyperthermophile Thermotoga maritima" by A. Grove and L. Lim (2001; J. Mol. Biol. 311, 491-502) who show "the unique DNA-binding properties of HU ... may be unique to HU from hyperthermophilic organisms." For Neutral Theory in the context of the genome of the malaria parasitge P. falciparum (Click Here) For PubMed Commentary (2015) Click Here [Sorry, the NCBI cut off links between commentaries and PubMed abstracts in March 2018. Some 7000 comments were salted away in a difficult to access files. Fortunately, a private site has them stored in more accessible form Click Here .] |
Szybalski's Transcription Direction Rule
|
|
Pyrimidine-rich and Purine-rich Clusters. Studies in Chargaff's laboratory resulted in methods to determine the concentrations of individual bases in DNA. Two sequences might have the same base composition, but differ dramatically in sequence. Prior to the emergence of exact sequencing methods, "cluster analysis" suggested a way of showing the uniqueness of individual sequences. This took advantage of the knowledge that, under acid conditions, DNA molecules readily loose purines (becoming "apurinic acids") and in these regions strand breakage is easily accomplished. Thus one is left with oligopyrimidine clusters wherever they happen to have existed in the molecule (e.g. CTTCT, CCCTCCT, TTCTTTTTTT, or generically, Yn). The types of clusters, and the frequencies of their occurrence, provided a unique "finger print" of the DNA molecule of origin. Of course, on a chance basis alone, one would expect some clusters. But it soon became apparent that the clusters were in much higher frequency than expected by chance. Thus, Chargaff (1963) in his Essays on Nucleic Acids summarized what we call his "cluster rule": |
"Another consequence of our studies on deoxyribonucleic acids of animal and plant origin, is the conclusion that at least 60% of the pyrimidines occur in oligonucleotide tracts containing three or more pyrimidines in a row; and a corresponding statement must, owing to the equality relationship, apply also to the purines." |
Clusters in Separate Strands of DNA
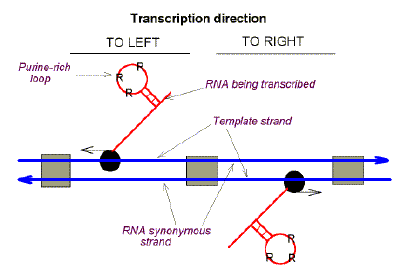
A major explanation for this abundance of clusters emerged from studies of the sedimentation properties of the separate strands of DNA. When the two strands of DNA were separated (this "melting" being achieved by heating), it was not intuitively obvious that the resulting single strands would differ in their densities to an extent allowing their separation on density gradients. Yet, this was observed with phage SP8 DNA by Marmur and Greenspan (1963: Science 142, 387). This appeared a lucky break for biochemists, allowing demonstration that one strand was used exclusively as a template for transcription in this organism. This "template-strand" was pyrimidine-rich (pyrimidines conferring greater density), and the other strand ("mRNA synonymous strand") was purine-rich (purines conferring less density). The generality of this became apparent from studies in Waclaw Szybalski's laboratory. Use was made of synthetic polyribonucleotides (usually containing only one base), which were hybridized at room temperature with natural single strands of DNA from various organisms. Normally, hybridization between two single stranded nucleic acid species is carried out at much higher temperatures with slow cooling over many hours (to achieve "renaturation" to duplex nucleic acid).
Whereas relatively short lengths of duplex SP8 phage DNA had been isolated intact, to isolate the long duplex strands from bacteria required a controlled degree of shearing producing a range of fragment sizes. These fragments were then heated to obtain the corresponding single strands. Fortunately, the chosen shearing conditions produced fragments usually containing one or a few genes. If the fragments had contained many genes, the base composition characteristics of genes transcribing one way would have cancelled out the base-composition characteristics of genes transcribing the other way. The phenomenon, which we now refer to as "Szybalski's transcription direction" rule (see the prescient Figure 4 of the following paper), would then have been much less apparent in the case of the large DNA molecules from bacterial sources. |
 |
Pyrimidine Clusters on the Transcribing Strand of DNA and Their Possible Role in the Initiation of RNA Synthesis
W. SZYBALSKI, H. KUBINSKI, AND P. SHELDRICK
McArdle Laboratory and Division of Neurosurgery, University of Wisconsin, Madison, Wisconsin
The Cold Spring Harbor Symposium in Quantitative Biology (1966) 31, 123-127 [Reproduced with the permission of W. Szybalski and Cold Spring Harbor Laboratory Press]
|
Little is known at present about the details of the molecular mechanism of the DNA-to-RNA transcription process, including some of its unique properties, as discussed in the preceding article: preferential initiation of RNA molecules with purine bases, and asymmetric copying of only one of the two DNA strands (Maitra, Cohen, and Hurwitz, this volume; Maitra and Hurwitz, 1965). Both of these features might be better understood if DNA molecules could be shown to contain on their transcribing strands some special base sequences serving as initiation points for RNA synthesis, and containing the pyrimidine bases complementary to the purines on the 5' termini of the RNA. Pyrimidine-rich clusters were observed by us and found to be asymmetrically distributed between the two strands of DNA isolated from a variety of bacteria, bacteriophages and higher organisms (Opara-Kubinska, Kubinski, and Szybalski, 1964; Kubinski, Opara-Kubinska, and Szybalski, 1966; Sheldrick and Szybalski, 1966). These clusters could be likely candidates for the specific initiation points of multi-cistronic RNA messages, perhaps by virtue of their high affinity for the DNA-directed RNA polymerase, and might correspond to the "promoter" sites deduced on the basis of genetic data (Jacob et al., 1964; Jacob, 1966). The techniques employed in detection of the pyrimidine-rich clusters in DNA were described earlier (Opara-Kubinska et al., 1964; Kubinski, et al., 1966), and will be only briefly outlined here. Deoxycytosine-containing clusters are revealed by the rapid complexing reaction between poly G or poly IG ribopolymers and single DNA strands containing such clusters. For example, when denatured DNA, extracted from B. subtilis cells or from the T1 or T7 coliphages, is mixed with poly G at temperatures between 4o C and 25o C, it separates into two fractions of different buoyant densities upon centrifugation in the CsCl density gradient (Fig. 1). The heavier (denser) fraction contains DNA strands complexed with poly G through their dC-rich sequences. The lighter (less dense) fraction comprises DNA strands that do not contain such dC sequences and cannot interact with poly G. When DNA of sufficiently high molecular weight is subjected to this fractionation procedure, each of the fractions comprises 1/2 of the total DNA and corresponds to one of the two complementary DNA strands, as shown by annealing experiments: each of the separate fractions cannot be self-annealed, whereas combining both fractions leads to renaturation of the DNA upon appropriate thermal treatment (4 hr, 65oC, 0.3 m Na+). Furthermore, it was found that the heavier fraction, which contains DNA strands characterized by dC-rich clusters, seems to comprise the majority of the RNA-transcribing strands, since it selectively hybridizes with pulse-labeled RNA (Kubinski et al., 1966; Sheldrick and Szybalski, 1966; Habich and Warner, 1966) and with ribosomal RNA as determined by Dr. K. Taylor of this laboratory.
|
|
|
|
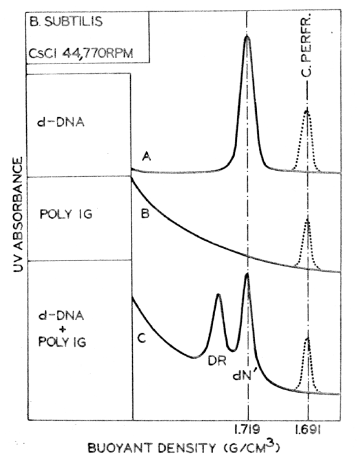
FIGURE I. Microdensitometric tracings of photographs taken after 22 hr of CsCl equilibrium density gradient centrifugation (44,770 rpm, 25oC) of denatured Bacillus subtilis DNA (A), poly IG (B), and a mixture of both (C). The heavier (denser) peak on the tracing C corresponds to the complex between poly G and one class of complementary DNA strands (left peak -- DR), whereas the peak to the right (dN') contains DNA strands that do not form complexes with poly G. The dotted line indicates the position of the density marker (native Clostridium perfringens DNA). |
|
The presence of dC-rich sequences on the transcribing DNA strand implies that corresponding dG-rich sequences should exist on the complementary, non-transcribing strand, and be detectable by their interaction with poly C. Such interactions were indeed observed, but only at a somewhat lowered pH when poly C began to be protonated (Sheldrick and Szybalski, in prep.). Since it is known that interstrand interactions in poly C depend on partial protonation of cytosine residues, one might conclude that detection of DNA-poly C interactions seems to be dependent on the formation of poly C aggregates, which in turn permit attaching sufficiently large amounts of poly C to the comparatively rare dG clusters on the non-transcribing strand of DNA. This lack of simple stoichiometry in the interactions between dC- or dG-rich clusters on DNA and complementary homoribopolymers does not permit a direct determination of the length of the dC clusters on the transcribing DNA strand. Resistance to ribonucleases and thermal stability data indicate that their length should be within 10 to 50 dC residues, probably with a few other bases randomly distributed within these cytosine runs. The shearing data indicate that there is one dC cluster per mol wt of one or a few million daltons. For the purpose of this discussion, the DNA strand characterized by predominance of dC sequences will be designated as the "C" strand or " - " (minus) strand. The minus sign indicates that in the several cases studied the C [Crick] strand was found to be transcribed into the mRNA (mRNA is usually designated as the "+" strand). The other DNA strand, which complexes with acidic poly C, will be designated as the "W" [Watson] or "+" (plus) strand (Figs. 2 and 3). This classification, which is admittedly oversimplified, does not pertain to the whole bacterial genomes but only to some phage genomes and to the fragments of bacterial DNA obtained by gentle extraction procedures. In addition to the dC and dG clusters, the DNA of most of the other organisms tested seemed to contain also dT- and dA-rich sequences (Fig. 2). The latter could be detected by measuring the interactions between denatured DNA and poly U. Unfortunately, no corresponding dT-poly A interactions have been hitherto observed under the limited number of experimental conditions evaluated in this laboratory.
|
|
|
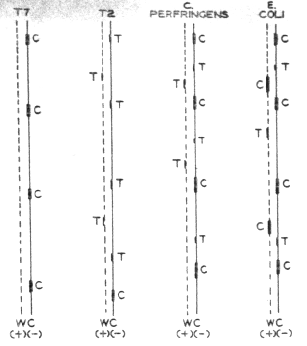
FIGURE 2. Diagrammatic representation of the distribution of dC-rich and dT-rich clusters in the DNA of four representative organisms. The complementary purine sequences are not indicated on the drawings. The strand exhibiting the predominance of pyrimidine-rich clusters and shown in some cases to correspond to the transcribing strand is drawn as a solid line (C, -). The other strand (W, +) is indicated by a dotted line. |
|
Some DNAS, however, such as the DNAs of the T3 and T7 coliphages, seem to contain only dC-rich clusters which apparently reside solely on one (transcribing?) strand, with the complementary dG sequences on the other strand (Fig. 2). Deoxyadenosine sequences and, by inference, dT clusters seem to be present on both DNA strands. In several cases, however (Clostridium perfringens, T2gt phage, and several Bacillus phages) a preponderance of dT-rich clusters was found on one of the DNA strands. In the case of Bacillus phages and C. perfringens, the same minus strand contains both dC clusters and a preponderance of dT clusters. Hardly any dC-rich clusters interacting with poly G were detected in T-even phage DNA (Fig. 2) or in the DNA of a few other species, including Cytophaga johnsonii. |
|
Although direct evidence is lacking, there are several indications that pyrimidine-rich clusters might correspond to the initiation points of RNA synthesis and, at the same time, determine which of the two DNA strands is being transcribed in a given operon. In small organisms, such as T7 coliphage, only one DNA strand throughout all its genome should be transcribed into RNA, since pyrimidine-rich clusters were observed only on one strand of the unbroken DNA molecule. In agreement with the genetic data indicating normal functioning of inverted sections of the bacterial chromosome, pyrimidine-rich clusters are observed also on the other DNA strand of several bacteria. One should remember, however, that opposite polarity of the complementary DNA strands would result in opposite directions of transcription. |
|
The indications that the pyrimidine clusters might correspond to the "promotor" regions (Fig. 3), which determine the initiation points of RNA transcription, are as follows:
|
|
|
|
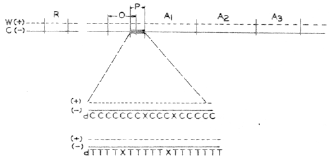
FIGURE 3. Molecular representation of the promotor region in the operon model. R, regulatory gene; 0, operater; P, promotor; A1 A2, A3, structural genes. The solid lines indicate the transcribing (C, -) strand of the DNA containing the pyrimidine-rich cluster in the promotor region, as indicated by the C (cytosine) or T (thymine) sequences. Letter X represents any of the other nucleotides. The nontranscribing (W, +) strand (dotted line) contains complementary purine-rich sequences in the promotor region. |
|
|
|
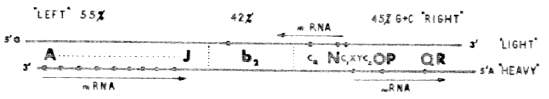
FIGURE 4. Working hypothesis for the distribution of the dC-rich clusters as related to the orientation of in vivo transcription of lambdaphage DNA (represented by arrows and mRNA symbols), based on the genetic and physical data discussed in this symposium (Hogness et al., Gros and Naono, Skalka, this volume; Eisen et al., Wu and Kaiser, as quoted by Hogness et al., this volume), and compatible with experiments summarized in this contribution, including the following preliminary data (W. S.) on the separation of the complementary lambda DNA strands. At the standard concentration of poly IG (20m g poly IG per 2 m g lambda DNA per 0.4 ml) the density increases (above that of the corresponding denatured DNA) amount to 5 and 16 mg/ cm3 (l c1b2+), 5 and 19 (l c1b2), and 8 and 10 (l dg(A-J), for the "light" and "heavy" strand, respectively. For the isolated left half (55% G + C) of the l c1b2+ DNA (prepared in Dr. A. D. Hershey's laboratory in Cold Spring Harbor in cooperation with Dr. A. Skalka) the corresponding density shifts are 1 and 24 mg/cm3, whereas there is little poly IG-effected separation of the strands derived from the right half (45 % G + C) of this DNA, with density increases of approximately 8 mg/cm3. The DNA strand which predominantly reacts with poly IG ("heavy") has 4 mg/cm3 lower density in alkaline CsCl gradients than the "light" strand (Added to proofs). |
|
When equating the pyrimidine-rich sequences in DNA, detected by DNA-polyribonucleotide binding, with RNA polymerase-binding, transcription-initiating "promoter" regions, we do realize that this correlation, even if true in principle, might be far from perfect. Our polynucleotide binding technique might be over-sensitive in some cases and not sensitive enough in others. We are especially apprehensive about quantitating the interactions between poly U and denatured DNA. Moreover, binding of polymerase need not imply obligatory initiation of the transcription process, even in the absence of any repressors. The only justifications for these speculations are the few suggestive correlations listed above and the otherwise complete lack of any molecular model for initiation of the DNA-to-RNA transcription, (Singer, and Leder, 1966), as related to the experimentally determined special features encountered in all naturally occurring, genetically active DNAs. |
SUMMARY Pyrimidine-rich clusters have been observed by us in DNA from a variety of organisms. In several cases it could be shown that the deoxycytidine clusters reside primarily, if not exclusively, on the mRNA-transcribing strand of the DNA. It appears also that a switch in the transcription from one to another complementary DNA strand is associated with the presence of deoxycytidine clusters in the transcribing regions of both strands. On the basis of these observations we propose a model in which such pyrimidine clusters introduce an element of discontinuity into the DNA helix by modifying its secondary structure, and serve as specific recognition sites for the initial binding of the RNA polymerase. In this manner they determine
When transcribing a given operon, all the mRNA molecules need not start at precisely the same point, as long as they include the initiating triplet for the first protein. Thus, such short DNA segments, with RNA polymerase-binding pyrimidine clusters on one strand and the complementary purine clusters on the other, might be regarded as physical equivalents of the "promotor" sites proposed on genetic grounds. |
|
ACKNOWLEDGMENTS. These studies were supported by a grant B-14976 from the National Science Foundation, by grant CA-07175 from the National Cancer Institute, U.S. Public Health Service, and by the Alexander and Margaret Stewart Trust Fund. REFERENCES CRAWFORD, L. V., E. M. CRAWFORD, J. P. RICHARDSON and H. S. SLAYTER. 1965. The binding of RNA polymerase to polyoma and papilloma DNA. J. Mol. Biol. 14: 593-597. GOLDTHWAIT, D. and F. JACOB, 1964. Sur le mecanisme de l'induction du developpoment du prophage chez les bacteries lysogenes. C. R. Acad. Sci., 259: 661-664. HARICH, A. and R. C. WARNER. 1966. Identification of the anticodon strand of B. megaterium DNA. Fed. Proc. 25: 707. HOWARD, F. B., J. FRAZIER, M. F. SINGER, and H. T. MILES. 1966. Helix formation between polyribonucleotides and purines, purine nucleosides, and nucleotides. II. J. Mol. Biol. 16: 415-439. HUANG, W. M., and P. 0. P. T'so. 1966. Physicochemical basis of the recognition process in nucleic acid interactions. I. Interactions of polyuridylic acid and nucleosides. J. Mol. Biol. 16: 523-543. JACOB, F. 1966. Genetics of the bacterial cell. Science 152: 1470-1478. JACOB, F., A. ULLMAN, and J. MONOD. 1964. Le promoteur, element genetique necessaire a 1'expression d'un operon. C. R. Acad. Sci., 258: 3125-3128. KUBINSKI, H., Z. OPERA-KUBINSKA, and W. SZYBALSKI. 1966. Patterns of interaction between polyribonucleotides and individual DNA strands derived from several vertebrates, bacteria and bacteriophages. J. Mol. Biol. 20: 313-329. MAITRA, U. and J. HURWITZ. 1965. The role of DNA in RNA synthesis, IX. Nucleoside triphosphate termini in RNA polymerase products. Proc. Natl. Acad. Sci. 54: 815-822. NEUHARD, J. and A. MUNCH-PETERSEN. 1966. Studies on the acid-soluble nucleotide pool in thymine-requiring mutants during thymine starvation. II. Changes in the amounts of deoxycytidine triphosphate and deoxyadenosine triphosphate in Escherichia coli 15 T-A-U-. Biochim. Biophys. Acta 114: 61-71. NISHIMURA, S., D. S. JONES, and H. G. KHORANA. 1963. Studies on polynucleotides. XLVIII. The in vitro synthesis of a co-polypeptide containing two amino acids in alternating sequence dependent upon a DNA-like polymer containing two nucleotides in alternating sequence. J. Mol. Biol. 13: 302-324. OPARA-KUBINSKA, Z., H. KUBINSKI, and W. SZYBALSKI.1964. Interaction between denatured DNA, polyribonucleotides, and ribosomal RNA: Attempts at preparative separation of the complementary DNA strands. Proc. Natl. Acad. Sci. 52: 923-930. SHELDRICK, P. and W. SZYBALSKI. 1966. Interaction of polyribonucleotides with the complementary strands DNA from certain Bacillus phages. Fed. Proc. 25: 607. SINGER, M. F., and P. LEDER. 1966. Messenger RNA: An evaluation. Ann. Rev. Biochem. 35: 195-230. T'so, P. 0. P., G. HELMKAMP, and C. SANDER. 1962. Interaction of nucleosides and related compounds with nucleic acids as indicated by the change of helix-coil transition temperature. Proc. Natl. Acad. Sci. 48: 686-698.
|
|
DNA Strand Asymmetries (Smithies et al. 1981). For more on Szybalski's transcription direction rule which, by virtue of
can provide information on the origin of replication and much more (Click Here).
For Szybalski's Transcription Direction Rule in Thermophiles Click Here |
| Sydney
Brenner (1991) |
 |
|
"Searching for an objective reconstruction of the vanished past must surely be the most challenging task in biology. I need to say this because, today, given the powerful tools of molecular biology, we can answer many questions simply by looking up the answer in Nature - and I do not mean the journal of the same name. ... In one sense, everything in biology has already been 'published' in the form of DNA sequences of genomes; but, of course, this is written in a language we do not yet understand. Indeed, I would assert that the prime task of biology is to learn and understand this language so that we could then compute organisms from their DNA sequences. ... We are at the dawn of proper theoretical biology." Evolution of Life. Ed. S. Osawa & T. Honjo. Springer-Verlag. |
|
Codon Choice and Grantham's Genome Hypothesis (1980) (Click Here)
Introns and Exons as a Way of Resolving Intragenomic Conflict (Click Here)
Thinking about Stem-Loops - Conceptual Problems (Click Here)
Chi Recombination Hot-Spots (Click Here)
Accounting Units in DNA (Click Here)
More on Chargaff's Second Parity Rule (Click Here)
Retroviral Speciation (Click Here)
Compositional Symmetry Principle (Click Here)
Trinucleotide Hierarchies (Click Here)
Mammalian-mode and Bacterial-mode Genome Organization in the Fruit Fly (Click Here)
Purine-Loading and Codon Choice in Thermophilic Bacteria (Click Here)
Double-Stranded RNA and Purine-Loading in Epstein-Barr Virus (Click Here)
Malaria Genome and Neutral Theory (Click Here)
Purine-Loading in Phage and Bacteria (Click Here)
Elementary Principles (Click Here)
Genome Conflict - Selfish Species Win (Click Here)
GC% as Recombinational Isolator (Click Here)
Amino Acids as Placeholders (Click Here)
Heredity as Information Transfer (Click Here)
Positive Darwinian Selection determined with a Single Sequence (Click Here)
Microsatellites Confer Extrusion Asymmetry (Click Here)
What is a gene (2009)? Click Here
Selfish Gene Revisited (2011) Click Here
Other Sites for Bioinformatics
For: Athenian Mathematical Biology from Aberdeen (Click Here)
For: Bioinformatics at the Rockefeller (Click Here)
For: Bioinformatics Course Online (Click Here)
For: Bioinformatics Textbook Online (Click Here)
For: Complexity (Click Here)
For: Cryptology at Oberlin College (Click Here)
For: DNA Computing (Click Here)
For: Gene Recognition and Characterization (Text CSHL) (Click Here)
For: Genomics Resources (Click Here)
For: Kropinski's On-Line Analysis Tools (Click Here)
For: Molecular Information Theory at NIH (Click Here)
For: Polish-style Bioinformatics (Click Here)
For: RNA Structure Analysis (Click Here)
![]()
Acknowledgements. Richard Salter of Oberlin College, Ohio, gave permission for the use of pictures from his fine Cryptology Web Page. Permissions for their articles and photographs were given by Andrew Ball, Erwin Chargaff, Richard Grantham, Vinayakumar Prabhu, Oliver Smithies, Noboru Sueoka, and Waclaw Szybalski.
![]()
BCHM/CISC-875*
Bioinformatics
Topics may include: DNA data analysis (genomics), secondary and tertiary structure analysis (nucleic acids and proteins), molecular scene analysis, evolutionary trees (phylogenetics), and computing with DNA. The course consists of:
Given jointly by the Departments of Biochemistry and of Computing and Information Science. Fall term; lectures and seminars. Coordinators
|
![]()
Some Bioinformatics Texts
R. F. Doolittle (1986) Of URFs and ORFs. A Primer on How to Analyze Derived Amino Acid Sequences. University Science Books.
G. von Heijne (1987) Sequence Analysis in Molecular Biology. Treasure Trove or Trivial Pursuit? Academic Pre ss.
M. Gribskov & J. Devereux (1991) Sequence Analysis Primer. Stockton Press.
M. S. Waterman (1995) Introduction to Computational Biology. Chapman Hall.
R. F. Doolittle (ed) (1996) Computer Methods for Macromolecular Sequence Analysis. Methods in Enzymology Volume 266. Academic Pre ss.
S. Schulze-Kremer (1996) Molecular Bioinformatics. De Gruyter.
J. Setubal & J. Meidanis (1997) Introduction to Computational Molecular Biology. PWS Pub. Co.
D. Gusfield (1997) Algorithms in Strings, Trees and Sequences: Computer Science and Computational Biology. Cambridge University Pre ss.
P. Baldi & S. Brunak (1999) Bioinformatics. The Machine Learning Approach. MIT Pre ss.
P. Pevzner (2000) Computational Molecular Biology: An Algorithmic Approach. MIT Pre ss.
A. Baxevenis & F. Ouellette (2001) Bioinformatics: A Practical Guide to the Analysis of Genes and Proteins. 2nd Edition. J. Wiley.
D. W. Mount (2001) Bioinformatics: Sequence and Genome Analysis. Cold Spring Harbor Laboratory Pr ess.
A. M. Lesk (2002) Introduction to Bioinformatics. Oxford Univ. Pre ss.
P. G. Young (2002) Exploring Genomes. W. H. Freeman.
D. R. Forsdyke (2006) Evolutionary Bioinformatics. Springer, New York. (Click Here)
D. R. Forsdyke (2011) Evolutionary Bioinformatics. 2nd Edition. Springer, New York. (Click Here)
![]()
Return to: Bioinformatics/Genomics Index (Click Here)
Return to: Evolution Index (Click Here)
Return to: HomePage (Click Here)
![]()
This page was established circa 1998 and last updated on 07 November 2020 by Donald Forsdyke.
WWW page access counterSince 30th March 1999