|
Differences and DNA Strand
Asymmetries in the Human Gg
and Ag Fetal Globin Gene
Region
Oliver Smithies, William R. Engels, John R. Devereux, Jerry L. Slightom and Shi-hsiang Shen
Laboratory of Genetics, University of Wisconsin, Madison, Wisconsin 53706
Cell 26,
345-353 (1981) Received June 11 1981: revised August 17, 1981
[With copyright permission from O. Smithies and Cell Press]
Base Substitutions versus Base Compositions
Length Differences
Interrelations between Length Differences and Base Substitutions
Regional Polarities and Strand Asymmetries
Chromosomal Domains Due to Strand Asymmetries
Chromosomal Domains in Other Systems
Biological Significance of Chromosomal Domains of Strand Asymmetry
| We have studied differences arising subsequent to the 5
kilobase pair (kb) duplication that led to the human Gg and Ag fetal globin genes. The local occurrence of base substitutions in
the duplicated 5 kb region correlates positively with the local AT base pair
content. This correlation also occurs in two mouse b-globin
genes and in two mouse immunoglobulin genes. The relationship is valid for transcribed or
nontranscribed DNA and for DNA that contains only coding sequences. Length differences in
the fetal globin duplicated regions correlate positively with the occurrence of short
direct repeats of ł 5 base pairs. Path analysis of the
interrelationships of base composition, base substitutions, repeats and length differences
provides an integrated view of the relative effects on chromosomal changes of these
variables and of selection. The distributions along the chromosome of simple sequences and of base compositions show highly significant local asymmetries between the transcribed and nontranscribed strands of the DNA, which permit us to divide the fetal globin gene region into chromosomal domains. Comparable domains are present in DNA from other sources, including the mammalian viruses SV40 and polyoma virus strain A-2 in which some of the domains appear related to discrete functions. |
In a previous paper (Shen et al., 1981), we presented, the complete nucleotide sequence of the human Gg and Ag fetal globin gene region. The data showed that a duplication of 5 kb of DNA led, about 34 million years ago, to the Gg and Ag genes, and that a subsequent gene conversion about 1 million years ago made the diverging regions virtually identical over 1.5 kb of their sequence. In this paper, we look at the differences between the duplicated regions and investigate their relationships to various features of the sequenced DNA. Examining the divergences that have occurred between these two relatively recently duplicated regions reveals some simple relationships between the occurrence of base substitutions and length differences in once identical regions of DNA and local properties of the DNA sequence.
We also find that the fetal globin region can be divided into relatively long chromosomal domains characterized by differing asymmetries in the base compositions of the transcribed and nontranscribed strands of the DNA. Comparable domains are present in DNA from several other sources, including the mammalian viruses SV40 and polyoma virus strain A-2 where some of the domains appear to be related to discrete biological functions.
Base Substitutions versus Base Compositions
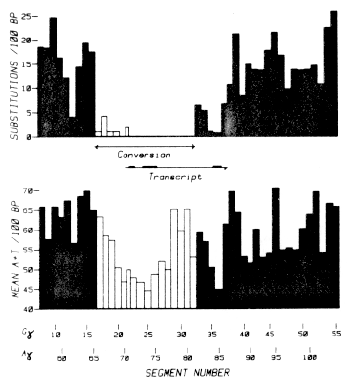
To analyze the distribution of various features along the 11,376 nucleotides of sequenced DNA from the human fetal globin gene region (Shen et al., 1981), we divided the region into 113 segments, each of approximately 100 nucleotides, and looked at the distribution of differences between the two 5 kb duplicated parts of the region. Differences between the duplicates due to base substitutions occurred more frequently in AT-rich segments than in GC-rich segments. Figure 1 presents the relevant data and shows that if we exclude the converted region, which has a different history from the rest of the DNA, the local variation in base substitutions along the region roughly parallels the local AT content of the DNA.
|
|
Figure 1. The Distribution along the Human Gg and Ag Fetal Globin Gene Duplication of Base Pair Substitutions and of A+ T Content. (Top) The base pair substitutions per 100 bp between the duplicated G g and Ag globin gene regions are shown along the sequenced DNA, which has been divided into numbered segments each of approximately 100 bp (see text). Segments 8 through 55 comprise the Gg duplicated region, and segments 57 through 103 comprise the Ag duplicated region. Segment 43 has no counterpart in the Ag region; it is omitted from the comparison, as are other (small) parts of either globin gene region that have no counterparts in the other gene region. The triply repeated element described by Shen et al. (1 981) is also omitted. (Bottom) The mean A+ T content per 100 bp is shown for each pair of segments along the duplicated region. For ease of visualization, bars representing data for the converted region (Conversion) are left unfilled. The transcribed region is indicated. |
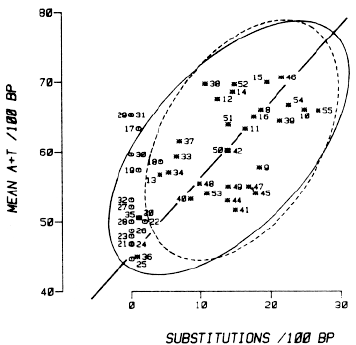
The relationship of base substitutions to local AT content is explored more fully in Figure 2, in which we plot percentage of substitutions against mean percentage of A+T content for the different chromosomal segments. The numbers associated with the data points in the main body of the plot refer to the segment numbers for the Gg part of the duplicated region. They permit each point to be located in the duplicated region by reference to Figure 1. For example, the numbered point 36 shows that segment 36, the third coding region in the Gg gene, differs from the corresponding segment in the Ag gene by 0.8% of its bases and that the two segments have a mean A+T content of 45%. The asterisk data points refer to segments outside the converted region of the duplication; the open points refer to segments within the converted region.
|
|
Figure 2. Substitutions per 100 Base Pairs between Paired Segments of the Gg and Ag Globin Gene Duplicated Regions versus the Mean A+ T Content of the Same Segments. The numbers identify the data points with respect to their (Gg ) segment numbers (see Figure 1). The 31 asterisk points refer to segments outside the converted regions. The 16 open points refer to segments within the converted region. The unbroken tolerance ellipse (see text) refers to the asterisk points. The dashed ellipse refers to the 26 segments in the nonconverted region, where no transcription is known to occur (Gg segments 8-16, 38-42 and 44-55). |
To provide a visual measure of the relationships between the variables, substitution and A+T content, we have drawn two ellipses in the figure. Both are 95% tolerance ellipses (see Diem and Lentner, 1970, p. 183). A 95% tolerance ellipse is such that, if a plot like that in Figure 2 were made with an infinite number of points from a bivariate normal distribution with the same degree of relationship as the present data, 95% of the points would fall within the ellipse. The unbroken tolerance ellipse refers to data from the 31 duplicated segments that have not been converted (that is, it refers to the 31 asterisk points, which in turn are represented by the filled bars of the diagrams in Figure 1). The dashed tolerance ellipse refers to a subset of the asterisk points to be discussed below.
To provide a numerical measure of the relationships between the two variables, we have used Kendall's t (Kendall, 1975). This statistic, which is based on the rank order of observations rather than on their magnitudes, tests the null hypothesis that if the observations are ranked in order with respect to one of the variables (neither takes precedence), then all permutations of order with respect to the other variable are equally probable. The test requires no assumptions concerning the underlying distributions of the variables. When t = +1 or -1, there is a perfect correlation between the two variables. When t = 0, there is no correlation. Using the methods described by Kendall (1 975), one can calculate a value from t for the probability, P, of obtaining under the null hypothesis a correlation at least as great as that observed.
The unbroken 95% tolerance ellipse in Figure 2 indicates visually that there is a significant relationship between the local occurrence of substitutions in the duplicated parts of the fetal globin gene region (excluding the converted portion) and the local A+T content. The value for t for these data (the 31 asterisk points) is 0.36, which corresponds to P = 0.0024; that is, the probability that 31 points drawn from two independent distributions would correlate this well is only 0.0024. Consequently, the relationship we are describing is very unlikely to be due to coincidences between two unrelated variables.
The data thus indicate that the number of base substitutions between the duplicated Gg and Ag gene regions at different localities along the duplication is related to the A+T content of these localities, with AT-rich regions having more base substitutions than GC-rich regions. For example, the third coding region, unit 36, has a particularly low AT content, and its percentage of substitution is almost zero. Likewise, the 31 end of the larger intervening sequence, unit 35, has a relatively low AT content and few substitutions. Units 33 and 34, which are from more internal but still unconverted parts of the large intervening sequence, have higher AT contents and more substitutions.
The dashed 95% tolerance ellipse in Figure 2 provides visual evidence that the relationship is still valid in those segments of the fetal globin duplication where no transcription is known to occur (again excluding the converted region). The Kendall t test likewise shows that there is still a significant positive correlation among these 26 segments (t = 0.29; P = 0.021).
The correlation between base substitutions and base composition raises the question whether the low (often zero) percentage of base substitutions in the 1.5 kb region we previously described as converted (Shen et al., 1981) is really due to conversion or whether the low percentage only reflects a low AT content in this locality. The question can be answered by determining whether the segments in the converted region (the 16 open points in Figure 2) are part of the same distribution as those in the unconverted region (the asterisk points). If they are, then the open points would be expected to lie with equal frequency below and above the major axis of the tolerance ellipse. If conversion has occurred, significantly more open points should lie above the major axis than below. In fact, 15 of the 16 open points lie above and only 1 below the major axis. The probability of this much or more deviation from equality occurring would be less than 0.0003 if the true distribution were binomial and symmetrical. The data thus show that those parts of the Gg and Ag genes corresponding to the converted region are more alike than would be expected from their base compositions, which supports our previous conclusions regarding the existence of this region of gene conversion.
We next asked whether this relationship between the two variables, base composition and base substitution, could be detected in the globin genes from a different species. The duplicated adult mouse globin genes, b Dmajor and b Dminor, and short stretches of their flanking sequences have been sequenced by Konkel et al. (1978, 1979). We aligned the 1394 comparable nucleotides from the two sequences and divided them into 1 4 segments of approximately 100 bases in the same way as we did for the Gg and Ag globin gene regions, omitting from the comparison any parts of either gene that could not be aligned with an equivalent part from the other gene. The correlation between the variables is again positive and significant (t = 0.45; P = 0.012), though the test is considerably less powerful this time, with only 14 instead of 31 segments of comparison.
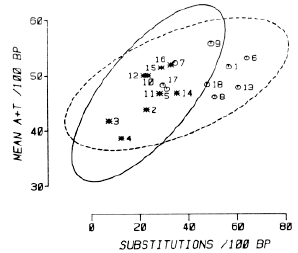
Finally, we asked whether the relationship of base composition to base substitutions could be detected in a pair of genes of a different category and, more stringently, whether it could be detected in exclusively coding sequences. The two mouse immunoglobulin heavy chain constant region genes, lgyl and lg-y2b, are closely related, as judged by their nucleotide sequences (Honjo et al., 1979; Tucker et al., 1979), and the coding nucleotide sequences of the three constant region domains in these two genes can be aligned with little ambiguity. This permits a comparison of nine segments, each approximately 100 bases in length. Figure 3 shows these data (asterisk points and unbroken ellipse) in a plot of mean AT content against base substitutions. Despite the small number of points, 9, the two variables are still significantly related with t = 0.54 and P = 0.023. Thus the relationship is detected even when relatively small amounts of nucleotide sequence that are strictly constrained by having to code for protein sequences of precise function are compared. (When all available data for the mouse g l and g 2b heavy chain constant region genes are included in the plot, the P value decreases to 0.014; see additional open points and dashed tolerance ellipse in Figure 3.)
|
|
Figure 3. Substitutions per 100 Base Pairs between Paired Segments of the Mouse Immunoglobulin Heavy Chain Constant Region Genes, Igg l and Igg 2b, versus the Mean A+T Content of the Same Segments The data points are identified by their segment numbers: (1) intervening sequence (IVS); (2, 3 and 4) CH1 coding sequence; (5, 6 and 7) IVS; (8) hinge region coding sequence; (9) IVS; (10, 11 and 12) CH2 coding sequence; (13) IVS; (14, 15 and 16) CH3 coding sequence; (17 and 18) 3' untranslated or flanking regions. The unbroken tolerance ellipse refers to the 9 segments that comprise the coding sequences for the 3 constant region domains: the dashed tolerance ellipse refers to all 18 segments. The DNA sequences are from Honjo et al. (1979) and Tucker et al. (1979). The homology alignment used in comparing the total sequences was given to us by F. R. Blattner. |
| These comparisons between diverging duplicated genes establish a relationship between local accumulation of base substitutions and local variations in the base pair composition of the DNA. The relationship appears general in that it applies to more than one class of gene, to nontranscribed or to transcribed DNA, and also to DNA that is transcribed and translated. The effect is relatively large, for it can result in tenfold differences in base substitutions in different localities along a chromosome. |
The biochemical basis for the observed relationship between base substitutions and local base composition is not likely to be revealed by the types of analysis used here. However, our analysis permits us to exclude some relatively trivial explanations of the relationship and to define its nature somewhat more closely. For example, to exclude the explanation that AT base pairs might simply be more likely to mutate than are GC base pairs, we counted the number of AT to TA substitutions in the fetal globin duplicated regions (we cannot distinguish the direction of these interchanges, but in either direction they involve mutations at an AT pair) and of GC to CG substitutions. We determined that the AT to TA substitutions do not occur more frequently than the GC to CG substitutions relative to the total numbers of AT and GC base pairs. Accordingly, our data are not explained by a simple difference in mutability of AT versus GC base pairs.
We also asked whether any of several other relationships between substitutions and local base composition might be more significant than the one we have detected. For example, we determined that local substitutions do not correlate significantly with local purine (or pyrimidine) content on either of the DNA strands. We also divided the substitutions into transversions and transitions. We found that both transversions ( t = 0.37; P = 0.0020) and transitions (t = 0.26; P = 0.021) are correlated with local AT contents, although the relationship is more extreme in the case of transversions.
We confirm Fitch's observations made with coding DNA (1980) that transitions occur more frequently than transversions and occur much more frequently than would be expected if all base pair changes were equally probable (we observed 278 transitions and 218 transversions versus expectations of 165 transitions and 331 transversions if all changes are equally probable). Thus the relationship of substitutions to AT content is more extreme for substitutions due to transversions than it is for substitutions due to transitions, despite the fact that transversions are relatively and absolutely less frequent than transitions. Possibly, locally AT-rich regions can accept base substitutions that distort the DNA double helix more easily than can locally GC-rich regions with their potentially higher melting temperatures.
The biological consequences of the relationship between substitutions and base composition are easier to understand. The relationship demonstrates that substitution rates are far from equal throughout the genome, and it suggests that local rates may have been adjusted by changes in local base compositions to levels appropriate for different regions. Regardless of whether such long term adjustments have occurred, the large regional variations in base substitutions that we have demonstrated are likely to be of considerable evolutionary importance.
We have previously pointed out (Efstratiadis et al., 1980; Shen et al., 1981) that differences in the lengths of duplicated globin gene regions are frequently associated with short direct repeats, typically of five or more nucleotides. This association is readily proved valid in a statistical sense. We previously noted (Shen et al., 1981) 32 length differences in the fetal globin gene duplicated region, all but one being less than 25 nucleotides. (The exception is 122 nucleotides in length.) We counted, in the duplicated regions of the chromosome, 271 independent direct repeats of 5 or more nucleotides having their first bases not more than 25 nucleotides apart. We then compared the distribution along the duplicated regions of these repeats and of the length differences (again omitting the converted regions) and found a significant positive correlation of these variables within the 31 nonconverted segments (Kendall's t = 0.36; P = 0.004). Thus there is a clearly established relationship between the occurrence of length differences and of short direct repeats.
The 1.5 kb converted region is demonstrably not part of the same distribution. There are 50 repeats in the converted region, and by comparison with the remainder of the duplication we would expect 5.9 length differences (5.9 = 50 x 32/271) if they occurred in strict proportion to the repeats. None was found. Assuming that length differences are Poisson distributed, we can reject the hypothesis that the converted region comes from a distribution with a mean of 5.9 at the significance level P = e –5.9 = 0.003.
Interrelations between Length Differences and Base Substitutions
When we compared the distribution of length differences with the distribution of base substitutions along the duplicated regions of the chromosome, we found that segments which have not diverged greatly by base substitution have usually not diverged by length changes either (Kendall's t = 0.29; P = 0.014 for 31 segments). Consequently, a fairly strong correlation exists between these two measures of divergence. We could think of no obvious reason why base substitutions should directly influence length differences or vice versa (although substitutions could act indirectly by increasing the frequency of repeats). However, an obvious additional variable, natural selection, could act coordinately on both base substitutions and length changes. For example, in a region where length differences are not selected against, base substitutions are likely to be of little importance; in a region where base substitutions are deleterious, length differences are also likely to be harmful. Thus selection would be acting in the same direction on both variables, and so they would be correlated.
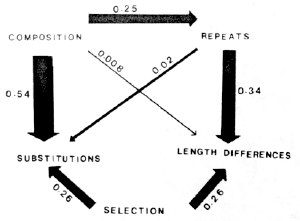
Path analysis (Wright, 1968) allows one to assess the relative importance of different causal effects among interrelated variables if these effects are assumed to be linear and a particular model (path diagram) is specified. One possible path diagram is shown in Figure 4, where the two measures of divergence, base substitutions and length differences, are considered to be dependent on all other variables. This figure illustrates the relative importance of the chosen paths by arrows with thicknesses roughly in proportion to their respective path coefficients, which are calculated by Wright's method from the correlation coefficients between all possible pairs of the variables.
|
|
| Figure 4. A Path
Analysis by the Method of Wright (1968) between the Five Indicated Variables. The several paths are illustrated by arrows with thicknesses roughly in proportion to their respective path coefficients (see text), which are given next to the arrows. |
Several other path diagrams were also studied, such as those with some of the directions of causation reversed, but the resulting path coefficients were all similar to those given in Figure 4, so that the listed path coefficients are robust. The path diagram in Figure 4 consequently provides an integrated view of the relative effects on chromosomal changes of the five variables: base composition, base substitutions, repeats, length differences and selection.
Regional Polarities and Strand Asymmetries
A study of the distribution along the chromosome of polynucleotide sequences (X)n, where n ł 5, revealed some unanticipated features. Most notably, some of these simple sequences are distributed nonrandomly along the chromosome and unequally between the two strands of the DNA.
| For example, on the 5' side of the transcribed region, poly(A) sequences occur much more frequently (13 times) on the codon-synonymous strand than do poly(T) sequences (4 times). Yet the reverse is true on the 3’ side, where poly(T) sequences occur more frequently (17 times) than poly(A) sequences (5 times). This is a highly significant difference (P = 0.0003 by Fisher's exact test). This polarity along the DNA in the occurrence of poly(A) versus poly(T) sequences implies that there are local asymmetries in the two DNA strands. |
We also examined the distribution along the codon-synonymous [mRNA synonymous] strand of repeated dinucleotides of the form (XY)n, or (YX)n, where n ł 3. With two exceptions, the frequency of occurrence of stretches of the six possible dinucleotides is in reasonable agreement with that predicted from the average frequency of the bases on the assumption that the DNA sequence is random.
One exception is that we expected 11.9 stretches of (AT)n and observed only 4; possibly there is some selection against the occurrence of poly(AT) stretches. The other, more unusual exception concerns the occurrence of stretches of the complementary dinucleotides, (AG) versus (TC) and (TG) versus (AG). We expected to find approximately equal numbers of these complements; however, in each case there is an excess of the G-containing dinucleotide relative to its complementary C-containing dinucleotide. For example, there are ten stretches of (AG)n, but only one stretch of (TC)n. (The probability of this much deviation from equality in either direction is 0.012 by the binomial test.) This observation again indicates an asymmetry between the two DNA strands and led us to investigate the base composition of the individual strands of the sequenced region.
|
|
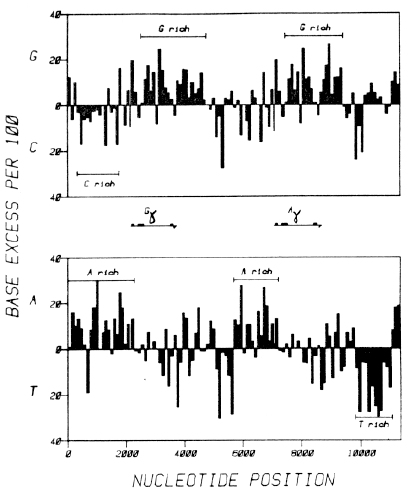
Figure 5. The Distribution over the Fetal Globin Gene Region of Asymmetries in the Base Composition of the Two Strands of Its DNA [Chargaff Difference Analysis]. The areas of the bars are directly proportional to the occurrence of whichever base is in excess of its complement ["Chargaff difference"]; their heights represent the excess of the indicated base normalized to 100 nucleotides. For example, segment 23, the first coding region of the Gg gene, is 92 nucleotides long and has the composition 33G, 15C, 28A, 16T; it has a normalized G excess of (33 - 15) x 100 / 92 = 19.6, and an A excess of (28-16) x 100/92 = 13.0. (Top) G versus C strand asymmetries. (Bottom) A versus T strand asymmetries. Data are presented for the codon-synonymous [mRNA-synonymous] strand. Numbers on the horizontal axis refer to nucleotide positions. Each bar corresponds to one of the 113 segments referred to in the text. The Gg and Ag transcribed and translated regions are indicated. Extent and location of six chromosomal domains (see text) of strand asymmetry are indicated. |
| Figure 5 shows the distribution of the four bases along the codon-synonymous strand of the sequenced DNA. The data are presented so that the excess of any base over its complement in each segment of the chromosome ["Chargaff difference"] is shown as a black bar of appropriate area. If there were no systematic strand asymmetries, the black bars for G excess and C excess (and for A excess and T excess) would be expected to occur randomly and with approximately equal frequency, and their total areas should be equal. These expectations are not met. There is obvious strand asymmetry in base compositions at various places in the sequenced region, and there are substantial local differences in these asymmetries along the chromosome. The differences are not due to differences of base pair composition (AT versus GC), which we presented in the lower part of Figure 1, but are due to differences in the occurrence of the bases on a single DNA strand (A versus T and G versus C). The A versus T asymmetries appear to be independent of the G versus C asymmetries. |
Chromosomal Domains Due to Strand Asymmetries
Inspection of the data plotted in Figure 5 enabled us to divide parts of the sequenced region into chromosomal domains. In each domain there is a marked excess of one of the bases relative to its complement. Until we understand the significance of these domains, assigning their boundaries is somewhat arbitrary. Nonetheless, the upper part of Figure 5 shows that there are two very G-rich domains, one in each of the duplicated regions. The two domains start approximately at the middle of the second coding region and extend over 2 kb, well past the poly(A) addition site. In both of these domains, there is a highly significant excess of the base G over its complement C. The Gg G-rich domain has 581 G nucleotides on the codon-synonymous strand and only 394 C nucleotides. This much deviation from equality in either direction is extremely improbable under the binomial distribution (P < 10-5). The Ag G-rich domain has a similarly improbable inequality of G versus C.
There is also a C-rich domain extending from close to the end of the Alu family repeat at nucleotide 335 to nucleotide 1700 (see Shen et al., 1981 for a discussion of this repeat). In this region, there are 296 C nucleotides and 218 G nucleotides (P < 10-3 under the binomial distribution). These tests establish unequivocally that there are highly significant asymmetries in the base compositions of the sense and antisense strands in these three regions. The data in the lower part of Figure 5 permitted us to recognize two A-rich domains and one T-rich domain.
The domains selected by inspection of the data in Figure 5 are the result of two factors, the existence of asymmetries in the base compositions of the two strands and the clustering of segments into groups (domains) of like asymmetry. We assessed the statistical significance of domains selected visually by considering a domain of length m (where m is the number of segments) in which n segments have an excess of one base over its complement (we call these "+" segments). We then calculated the probability according to two null hypotheses that a domain of this (preselected) length m with at least n "+" segments would be found by chance in a linear sequence of s segments. Null hypothesis (i) assumes that " + " and " - " segments are equally probable and uniformly distributed; it tests for significant domains assuming that the two DNA strands should be symmetrical. Null hypothesis (ii) assumes that " + " and " - " segments are uniformly distributed but not necessarily equally probable; it tests for significant domains even if strand asymmetries are accepted. Details of the calculation are given in Experimental Procedures. The resultant probabilities provide a measure of the significance of the domains we are describing, The probabilities for the six domains in Figure 5 clockwise from top left are 0.051, 0.0021, 0.0068, 0.0096, 0.061 and 0.0038 by null hypothesis (i), and 0.0039, 0.048, 0.10, 0.0026, 0.l7 and 0.019 by null hypothesis (ii). These low probabilities indicate significant local variations in the strand asymmetries along the length of the sequence, irrespective of whether or not strand asymmetries are accepted in the sequence as a whole.
The known coding sequences in the fetal globin gene region make very small contributions to the observed strand asymmetries. Accordingly, the domains are unlikely to result from specific codon requirements for the Gg and Ag globin polypeptides.
Chromosomal Domains in Other Systems
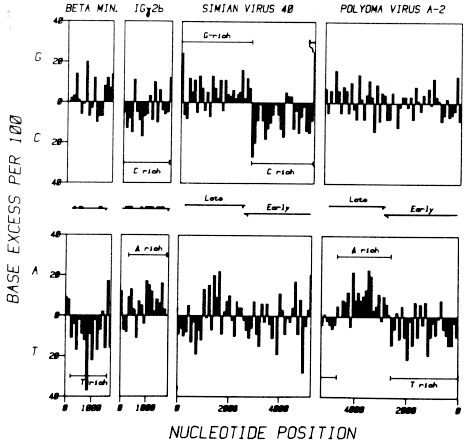
We have looked for chromosomal domains due to strand asymmetries in base composition in several other sequenced eucaryotic DNAS, including the mouse b minor globin gene (Konkel, 1979), and the mouse immunoglobulin g2b heavy chain constant region gene (Tucker et al., 1979), and two mammalian DNA tumor viruses, SV40 (Reddy et al., 1978; Fiers et al., 1978) and polyoma virus strain A-2 (Friedmann et al., 1979; Deininger et al., 1979; Griffin et al., 1980; Tooze, 1980). In each case, we find marked strand asymmetries and strong evidence for chromosomal domains. Figure 6 is a plot of these data. The DNA for the mouse b minor globin gene is markedly T-rich over the transcribed region on the codon-synonymous strand; P = 0.0024 on null hypothesis (i). The mouse immunoglobulin g 2b heavy chain constant region gene, however, is A-rich (P = 0.015) and C-rich (P = 0.033). It appears that completely different strand biases can be found in different systems.
|
|
Figure 6. The Distribution of Asymmetries in the Base Composition of the Two Strands of Several Mammalian DNA Sequences. The areas of the bars are proportional to the occurrence of whichever base is in excess of its complement. The diagrams are for mouse adult bminor globin (Konkel et al., 1980); mouse immunoglobulin g2b heavy chain constant region (Tucker et al., 1979); simian virus 40 (Reddy et al., 1978; see also Fiers et al., 1978; Tooze, 1980); and mouse polyoma virus strain A-2 (Friedmann et al., 1979; Deininger et al., 1979; Griffin et al., 1980). We used the DNA sequence numbering system of Reddy et al. (1978) for SV40, with the 17 nucleotide correction (Tooze, 1980); the polyoma virus numbering system is in the reverse orientation, but the data are presented in the figure so that they are comparable. The transcribed regions are indicated between the upper and lower bar diagrams, which refer to G versus C and A versus T strand asymmetries ["Chargaff differences"]. |
| A comparison of the strand asymmetries and chromosomal domains of the two well characterized viral genomes is particularly informative. The circular SV40 genome is obviously divisible into two domains. A G-rich domain extends from nucleotide 5201 (-43) to nucleotide 2700 (P = 0.016); a C-rich domain extends from nucleotide 2701 to nucleotide 5200 (P =0.0040). The genome of the polyoma strain A-2 virus is also divisible into two domains on the basis of strand asymmetries (P = 0.0048 and P = 0.056), but the domains in polyoma are due to A versus T asymmetries rather than to the G versus C asymmetries seen in SV40. This is surprising, since the two viruses show considerable homology and have many functional similarities (Tooze, 1980). However, the domains in both viruses share the one striking feature that their boundaries appear to correspond fairly closely to the transcriptional boundaries of the two genomes; the domains are also all purine-rich on the codon-synonymous strand. |
Biological Significance of Chromosomal Domains of Strand Asymmetry
| The data we have reviewed from over 25 kb of eucaryotic DNA show that strand asymmetries in base composition are of general occurrence. The asymmetries allow the recognition of chromosomal domains that are often over 2 kb in length. Domains may be rich in any of the four bases. G versus C and A versus T domains appear to be possible in a single DNA region. In some cases, the domains coincide fairly well with known biological functions. Yet similar functions (including the coding of homologous proteins) can be realized in spite of different strand asymmetries. It is as if selection can produce a variety of strand asymmetries that are still compatible with the underlying demands imposed by transcription and coding of proteins. |
Strand asymmetries in base composition are not unique to eucaryotic DNA. Over 15 years ago, Szybalski and his colleagues (1966) described the asymmetric occurrence of pyrimidine clusters on the strands of DNA isolated from a variety of bacteria and bacteriophages. The authors noted that pyrimidine richness predominated on the transcribed strand (which corresponds to purine richness on the codon-synonymous strand). Our data show that the transcribed regions of the human fetal globin genes and of the two viruses are also purine-rich on the codon-synonymous strand. This predominance is not, however, universal, since the mouse b minor globin gene is pyrimidine-rich and the mouse Igg 2b gene is neither purine-rich nor pyrimidine-rich.
We looked for domains due to strand asymmetry in two sequenced regions of procaryotic DNA: 4.4 kb from the E. coli plasmid pBR322 (Sutcliffe, 1979), and 3.4 kb from the trp operon of E. coli (Nichols and Yanofsky, 1 979: Christie and Platt, 1980; Crawford et al., 1980). We found a T-rich domain (P = 0.0064) in pBR322 extending clockwise about 1.2 kb from the beginning of the tetracycline resistance genes, but found no domains in the trp operon sequence.
| Our studies of the distribution of base pair substitutions and length differences in the duplicated portions of the human fetal globin gene regions and in other duplicated genes have provided us with an integrated view of the interrelationships of various changes that can alter the DNA of a chromosome. They show that the evolutionary changes of a local region of the genome can correlate with gross features of its sequence, such as local base compositions, which are at least partly independent of specific functions. Studies of the distribution of simple sequences and strand asymmetries in the human fetal globin gene region and other eucaryotic DNA sequences have led us to recognize relatively large-scale chromosomal domains having different strand asymmetries, but we do not know their biological significance. |
Statistical Tests for Domains
The following analysis was used to identify regions of DNA (domains) in which one of the two complementary bases of a given kind of base pair predominates significantly. Each of the regions we selected by inspection of the data in Figures 5 and 6 was considered to be a candidate for such a domain. We then asked whether the composition of the candidate domain is unusual given the length of the sequence from which it was selected and and the null hypothesis of either uniform and equal frequencies (i) or uniform but not necessarily equal frequencies (ii) of the two complementary bases.
Suppose a candidate domain of length m (number of segments) is selected from a sequence of total length s because of an excess of one base (" + ") over its complement (" – ") in n segments. We want to calculate under our two null hypotheses
P = Prob{max(X1, X2,…,Xs-m+1) ł n} (1)
where Xi is the number of " + " segments (that is, segments with an excess of " + ") in the m contiguous segments beginning with segment i, and n is the observed number of " + " segments in the candidate domain. The probability thus calculated may be considered as a significance level in tests for the existence of a domain. In interpreting this significance level, it should be noted that, although the length m of the candidate was determined subjectively (by inspection of the data in Figures 5 and 6), the test takes account of the number of opportunities (related to s) for choosing a candidate of length m.
The probability distribution of the X vector was calculated with a model in which each segment of the sequence is chosen independently of every other segment. For every segment, the probability of being a " + " segment is a constant value p. Under null hypothesis (i) p = 0.5, and under null hypothesis (ii) p is set to the observed proportion of "+ ' segments in the entire sequence under study. X1 was thus assumed to come from a binomial distribution with index m and parameter p. To get each subsequent Xi , one of the segments in the current X is discarded at random and a new segment is chosen. The resulting stochastic process, commonly known as a birth-and-death Markov process, can then be handled by standard methods (Feller, 1950). If Xi reaches the value n, that particular Markov chain is terminated. Accordingly, in calculating P, we may consider Xi = n to be an absorbing state, so that the transition matrix, A, is
| A= | ip/m + (m-i)(1-p)/m | 0< or = j < n |
| i(1-p)/m | 0< or = j - 1 < n | |
| (m -i)p/m | 0<i = j + 1 < or = n | |
| 1 | i = j = n | |
| 0 | otherwise |
P = Sum from i = 0 to n of b(i|m, p) Ani(s-m)
in which Ani(s-m) is an element in As-m and b(i|m, p) is the binomial probability of i for i = 0, 1, 2, . . . , n - 1, and b(n|m, p) = 1 - b(0|m, p) - … - b(n - 1 |m, p). For circular genomes we use Ani(s) in place of Ani(s-m) in equation (3).
Acknowledgments
We would like to thank Roland Littlewood and Paul Haeberli for assistance with the computer analyses, and Walter Fitch for extremely helpful discussions of our ideas. This work was supported by grants from the National Institutes of Health, and this paper is number 2503 from the Laboratory of Genetics, University of Wisconsin.The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. Section 1 734 solely to indicate this fact.
References
Christie, G E. and Platt. T (1980). Gene structure in the tryptophan operon of Escherichia coli. J. Mol. Biol. 142. 519-530.
Crawford, I. P., Nichols, B. P. and Yanofsky, C. (1980). Nuclectide sequence of the trpB gene in Escherichia coli and Salmonella typhimurium. J. Mol. Biol. 142, 439-502
Deininger. P., Esty A., LaPorte. P. and Friedmann, T (1979). Nucleotide sequence and genetic organization of the polyoma late region: features common to the polyoma early region and SV40 Cell 18, 771-779.
Diem. K. and Lentner, C (1970). Documenta Geigy. Scientific Tables. (Ardsley. New York: Geigy Pharmaceuticals), p. 183.
Efstratiadis, A., Posakony, J. W., Maniatis, T., Lawn, R. M., 0’Connell, C, Spritz, R. A.. DeRiel, J. K., Forget, B. G., Weissman, S. M., Slightom, J. L., Blechi, A. E , Smithies, O., Baralle, F. E., Shoulders, C. C. and Proudfoot, N. J. (1980) The structure and evolution of the human b-globin gene family. Cell 21. 653-668.
Feller. W (1950) An Introduction to Probability Theory and Its Applications. (New York: John Wiley and Sons).
Fiers W., Contreras. R., Haegeman, G., Rogiers, R., Van de Voorde, A. Van Heuverswyn, H., Van Herreweghe, J., Volckaert. G. and Ysebaert, M (1978). Complete nucleotide sequence of SV40 DNA. Nature 273, 113-120.
Fitch, Walter M. (1980). Estimating the total number of nucleotide substitutions since the common ancestor of a pair of homologous genes: comparison of several methods and three beta hemoglobin messenger RNAs. J. Mol. Evol. 16, 153-209.
Friedmann, T., Esty, A., LaPorte, P. and Deininger, P. (1979). The nucleotide sequence and genome organization of the polyoma early region: extensive nucleotide and amino acid homology with SV40. Cell 17, 715-724.
Griffin, B. E., Soedo, E., Barrell, B.G. and Staden, R. (1980). Sequence and analysis of polyoma virus DNA. In DNA Tumor Viruses. J. Tooze, ed. (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory), p. 831.
Honjo, T., Obata, M., Yamawaki-Kataoka,Y., Kataoka, T., Kawakami, T., Takahashi, N. and Mano, Y. (1979). Cloning and complete nucleotide sequence of mouse immunoglobulin g l chain gene. Cell 18, 559-568.
Kendall, M. (1975). Rank Correlation Methods. (London: Charles Griffin and Company Ltd.), Chapter 3.
Konkel, D. A., Tilghman, S. M. and Leder, P. (1978). The sequence of the chromosomal mouse b -globin major gene: homologies in capping, splicing and poly(A) sites. Cell 15, 1125-1132.
Konkel, D. A., Maizel, J. V., Jr., and Leder, P. (1979). The evolution and sequence comparison of two recently diverged mouse chromosomal b -globin genes. Cell 18, 865-873.
Nichols, B. P. and Yanofsky, C. (1 979). Nucleotide sequences of trpa of Salmonella typhimurium and Escherichia coli: an evolutionary comparison. Proc. Nat. Acad. Sci. USA 76, 5244-5248.
Reddy, V. B., Thimmappaya, B., Dhar, R., Subramanian, K. N., Zain, B. S., Pan, J., Gosh, P. K., Celma, M. L. and Weissman, S. M. (1978). The genome of simian virus 40. Science 200, 494-502.
Shen, S.-H., Slightom, J. L. and Smithies, 0. (1981). A history of the human fetal globin gene duplication, Cell 26, 191-203,
Sutcliffe, J. G. (1 979). Complete nucleotide sequence of Escherichia coli plasmid pBR322. Cold Spring Harbor Symp. Quant. Biol. 43, 77-90.
Szybalski, W., Kubinski, H. and Sheldrick, P. (1966). Pyrimidine clusters on the transcribing strand of DNA and their possible role in the initiation of RNA synthesis. Cold Spring Harbor Symp. Quant. Biol. 31, 123-127.
Tooze, J., ed. (1980). DNA Tumor Viruses. (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory).
Tucker, P. W., Marcu, K. B., Newell, N., Richards, J. and Blattner, F. R. (1979). Sequence of the cloned gene for the constant region of murine g 2b immunoglobulin heavy chain. Science 206, 1303-1306.
Wright, S. (1968). Evolution and the Genetics of Populations, 1. Chapter 13. (Chicago, Illinois: University of Chicago Press).
For purine-loading in thermophiles: (Click Here)
Return to: Bioinformatics Index (Click Here)
Return to: Evolution Index (Click Here)
Return to: HomePage (Click Here)
Established circa 1999 and last edited 04 Sep 2010 by Donald Forsdyke