|
|
|
|
|
|
|
MALARIA
|
|
|
|
|
|
|
|
| Malaria is caused by one of four species of Plasmodium (falciparum, vivax, malariae and ovale). Of these P. falciparum is the most lethal being estimated to cause 200 million clinical cases, and 1-3 million deaths (including many children) every year world-wide. The first of the papers below deals with the strangely low rate in P. falciparum of the type of mutation that does not change the encoded amino acids ("synonymous mutations"). The second paper deals with the mysterious low complexity segments that are found in many P. falciparum proteins. |
 |
![]()
![]()
Review: Trends in Parasitology (2002)
18,
411-418
With copyright permission from Else vier Science.
Selective pressures that decrease synonymous mutations in Plasmodium falciparum
Donald R. Forsdyke
![]()
![]()
Classical neutral theory implies that absence of non-amino acid changing mutations in P. falciparum results from a recent population bottleneck. However, absent mutations can also reflect conservation of the genome phenotype. Keywords: Plasmodium falciparum, synonymous mutation, positive selection, neutral hypothesis, low complexity segments, GC-pressure, AG-pressure, fold pressure
Rich and Ayala propose that the zero rate of non-amino acid changing (synonymous) mutations in some proteins of Plasmodium falciparum reflects a recent population bottleneck. Alternatively, Arnot and Saul propose sequence conservation in response to selective pressures other than the pressure to encode protein. Among these are fold pressure and purine-loading pressure. Genomes adapt to these by acquisition of introns and/or low complexity (simple sequence) segments in proteins. Adaptive explanations include facilitation of intragenic recombination (and hence diversification of the encoded protein) by DNA stem-loop secondary structures. |
![]()
Negative and positive selection are two, mutually exclusive, consequences of a base mutation in a nucleic acid sequence. The extreme imperative of negative selection is: if you mutate, you die. The extreme imperative of positive selection is: if you do not mutate, you die. Occupying the middle-ground between these extremes are mutations that may lead to either weak positive or weak negative selection. In these cases, there will be long-term effects on the number of progeny. The existence and size of a class of mutations that are truly neutral, in that there are no long-term effects on number of progeny, are subjects of much debate1-8. Whether a base mutation will lead to negative or positive selection depends on its location. A mutation affecting the active site of an enzyme will usually disturb enzyme function and this may impair the function of an organism to the extent that it will produce fewer descendents than will organisms without the mutation (i.e. its fitness to reproduce its kind is impeded). In the extreme, this is ensured by the death of the organism that has the mutation. On the other hand, a mutation affecting an antigenic determinant at the surface of a pathogen may allow it to evade the immune defences of its host. A pathogen mutating in this way may produce more descendents than will pathogens without the mutation (i.e. its fitness to reproduce its kind is enhanced). In the extreme this is ensured by the death of pathogens that do not have the mutation. If selection is not balanced (e.g. in the case of major histocompatability complex polymorphism), this may generate a population bottleneck (i.e. decreased population diversity). |
Measurement of negative and positive selection
Nucleic acid bases that are conserved among related organisms (i.e. are evolving slowly) are likely to affect functions subject to negative selection, and nucleic acid bases that are not conserved among related organisms (i.e. are evolving rapidly) are likely to affect functions subject to positive selection. Thus, a determination of evolutionary rate can assist the distinction between bases under positive selection, and bases under negative selection. For this, intraspecies or interspecies base differences between organisms can be determined and calibrated against some temporal scale. Accumulation of a large number of differences in a short period would indicate positive selection. However, accurate temporal calibration is difficult. An amino acid can usually be encoded by more than one nucleic acid codon (termed 'synonymous' codons). A mutation changing a codon to a non-synonymous codon will, by definition, change the encoded amino acid. To provide a relatively time-independent, internal, frame of reference for determining the form of selection, it has been found convenient locally to compare non-synonymous mutations per non-synonymous site (dn) to synonymous mutations per synonymous site (ds). This assumes that synonymous mutations are adaptively neutral, and hence reflect the 'background' rate of accepted (uncorrected) mutations in the genomic segment of interest3. Thus the dn/ds ratio within a nucleic acid segment would to provide an index of the rate at which that segment is evolving. A high ratio would suggest that the segment is under positive selection. A low ratio would suggest that the segment is under negative selection. |
|
In most cases ds values are significantly above zero, and determinations of dn/ds agree with expectations. However, for certain proteins of the malaria parasite, Plasmodium falciparum, ds values are at, or close to, zero. Rich and Ayala9-13 interpreted this as revealing a population bottleneck following which there had been insufficient time for mutations to accumulate. Thus, at the extreme, existing species members could have derived from one recent 'Eve' . However, Arnot14-15 and Saul16-17 argued that zero ds values could result from high conservation of bases that do not determine the nature of an encoded amino acid (a violation of the 'neutral' assumption). In this case, both the recent origin argument, and calculations based on dn/ds ratios could be invalid. The recent origin argument was also contested by Hughes and Verra18-20. However, the finding of few mutations in certain non-protein-encoding regions (introns) was held by Volkman et al.21 to support the argument. Both parties concede conceptual problems and the situation is described as 'paradoxical'22. The purpose of the present communication is to introduce new evidence supporting the argument of Arnot and Saul. It is suggested that the genome of P. falciparum is more sensitive than other genomes to several non-classical selective factors, which collectively constitute the 'genome phenotype'. |
|
There are selective pressures on nucleic acid sequences other than the pressure to encode proteins and certain RNAs1. This led to Grantham's 'genome hypothesis' in 198023, and the recognition that, distinct from the classical phenotype, there is a 'genome phenotype'24 moulded by 'forces that act only on the level of individual nucleotides'25. Some of these forces involve groups of bases, the symbols for which are shown in Table 1 (e.g. G and C are referred to collectively as S). Some groups are quantified as the sum of two bases in a segment (e.g. S = G + C). Others are quantified as the difference between two bases in a segment: (e.g. ∆S = G - C). |
|||||||||||||||
|
|||||||||||||||
|
Several non-classical pressures on genome sequences are listed in Table 2. Saul17 rested his arguments primarily on GC-pressure and pressures acting at the translation level. GC-pressure, AG-pressure and fold pressure will be considered here. GC-pressure might be referred to as S-pressure, but GC-pressure has acquired popular usage and will be used here. Similarly, AG-pressure (purine-loading pressure) might be referred to as R-pressure but, for consistency, the two-letter designation is used here. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
The pressures are not independent either of each other, or of classical phenotypic pressures. For example, as GC-pressure increases, base C can be acquired at the expense of base A, so AG-pressure decreases26,27. Very small changes in S bases (assessed as the percentage of G + C in a segment of duplex DNA) can have a major impact on fold pressure28. GC-pressure influences protein composition29 and extreme values (low or high) can correlate with decreases both in observed synonymous mutations30 and in protein complexity31. While the adaptive roles of pressures affecting the genome phenotype may be controversial, of their existence there is little doubt5. Thus, a sequence that is evolving slowly with respect to bases that do not determine amino acids could be under negative selection with respect to these pressures (i.e. in the extreme, an organism that mutates these bases dies, and in protein-encoding regions ds values are kept low). That the genome of P. falciparum might be highly susceptible to such non-amino-acid-changing pressures was evident in a recent study by Pizzi and Frontali32 of low complexity (simple sequence) regions in P. falciparum proteins (see below). |
Long considered a manifestation of biases in cellular mutational mechanisms29, the case for a selective role of GC-pressure is now stronger. Small differences in percentage GC should suffice to impede pairing at meiosis, and initiate species divergence5, 28, 33, 34. Consistent with this, Saul17 has proposed that, in the genus Plasmodium, high substitution rates would characterize speciation events. Following this, species GC integrity would be sustained by selection. Thus, co-infection of a single mosquito by two species would be mutually inhibitory due to impaired meiosis, and eventually fewer and/or less fit sporozoites would emerge. In this way allied species exert a selection pressure on each other's percentage GC8. |
|
When compared with their homologs in other organisms, most proteins of P. falciparum are longer by virtue of their content of low complexity regions that include internally-repetitive runs of amino acids (tandem arrays, homopolymer runs). Such low complexity regions are in non-globular domains of 90% of the open reading frames in chromosomes 2 and 335, 36. The observed amino acid preferences of these regions correlate, not with the properties individual amino acids confer (e.g. hydrophilicity, volume, flexibility), but with the A-richness of their codons. Accordingly, Pizzi and Frontali32 concluded that there is 'an amino acid choice strongly conditioned by a genome property (i.e. A-richness in the coding strand)'. The pressure to purine-load the mRNA-synonymous strands of DNA was noted in a bacteriophage in 196637 and, apart from a few intriguing exceptions, is likely to be a general phenomenon (reviewed in Refs 5-8). Similar to GC-pressure, AG-pressure can sometimes dominate the amino acid content of proteins (i.e. the needs of the genome phenotype are served better than those of the classical phenotype26, 27). Evaluation of AG-pressure as the purine-loading index (PLI) is outlined in Table 3. |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
The needs of secondary structure can sometimes dominate the sequence and composition of a nucleic acid38-39. The well-known ability of single-stranded tRNA molecules to fold into complex structures is reflected in the potential of the corresponding duplex DNA sequences (tRNA genes) to adopt similar structures when the two strands of the duplex separate. The stability (folding energy) of such structures is significantly greater than that of the structures generated from the same sequences after base order is shuffled. In the course of evolution, the order of bases appears to have been arranged to maintain the stem-loop configurations that contribute to the overall structure. Thus, the cytoplasmic RNAs that function by virtue of their structure [e.g. tRNAs and ribosomal RNAs (rRNAs)] rather than by encoding information for amino acid sequences [messenger RNAs (mRNAs)], are transcribed from DNA sequences with a similar potential for structure when in single-stranded form. However, many mRNAs also show the ability to adopt significant higher ordered structures and this is reflected in the potential for structure of the corresponding DNA sequences. That the structure of mRNAs might not necessarily relate to their own functioning became evident when it was observed that a similar or greater potential for higher ordered structure is found in introns and intergenic regions. The potential to form stem-loop structures of significantly greater stability than that of the corresponding randomized sequences is a general property of biological DNA sequences40-43. The evaluation of the relative contributions of base composition and base order to the folding energy of a DNA segment is outlined in Table 4. |
||||||||||||||||||||||||
|
||||||||||||||||||||||||
|
When the needs of the genome phenotype (e.g. GC-pressure, AG-pressure, fold-pressure) are not compatible with the needs of the classical phenotype (e.g. amino acid composition and sequence of a protein), there is a conflict which appears to be ameliorated by the insertion of introns40,41,43 and/or of simple sequence elements. The gene encoding Epstein-Barr Nuclear Antigen-1 (EBNA-1) of Epstein-Barr virus (EBV) is instructive in this respect27. EBV has various forms of latency in its human hosts, a state associated with the transcription of only a few 'latency-associated transcripts'. All forms of latency have in common the transcript encoding the EBNA-1 protein. This contains a long simple-sequence element (diagonally striped box in Fig. 1) that can be removed without interfering with known protein functions27. Because the EBV genome appears to be under pressure for compactness (i.e. there are few introns and little intergenic DNA), the simple-sequence element should only be present if, under some circumstance, it confers an adaptive advantage either at the protein level, or at the level of the encoding nucleic acid. One function of the element is to facilitate the purine-loading of the corresponding mRNA (Fig. 1a)27. In the EBV genome (60% GC), purines are contributed both by A (measured as DW) and by G (measured as DS). In organisms with a very low GC%, such as P. falciparum, A is the major contributor to the PLI (see below).
|
|
|
|
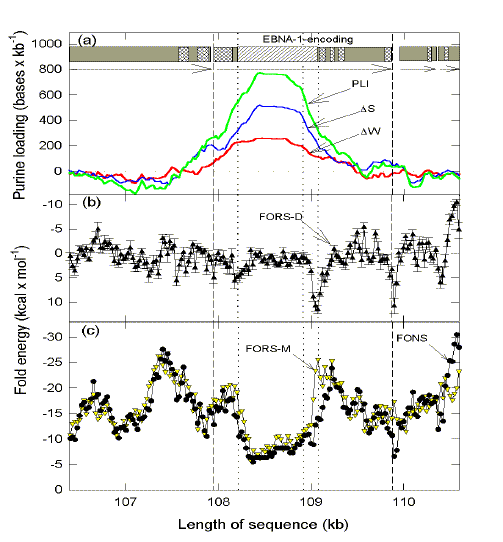
Fig. 1. Chargaff difference ('skew') analysis
(a) and fold analysis (b,
c) of part of the Epstein-Barr virus
(EBV) genome encoding the EBNA-1
antigen (GenBank V01555). Alignments of exon boundaries are indicated by
vertical dashed lines.
In (a) open reading frames and transcription directions are shown as grey boxes with horizontal arrows. The region encoding the low complexity simple-sequence repetitive element in EBNA-1 is shown as a white box with diagonal stripes; vertical dotted lines indicate its division into segments encoding mainly Gly-Ala repeats (left) and Gly-Arg repeats (right). Other low complexity elements are shown as cross-hatched white boxes. PLI values (green line) are the sum of ∆W values (red line) and ∆S values (blue line). In (b), values for the base order-dependent component of the folding energy (FORS-D) are shown with standard errors. In (c), values are shown for the base composition-dependent component of the folding energy (FORS-M), and for the total folding energy (FONS). Symbol and line designations are indicated by sloping arrows. Data points are either at the centre of 400 base windows (overlapping by 25 bases in (a)), or at the centre of 200 base windows (overlapping by 25 bases in (b) and (c)). Data are adapted from Ref. 27 updated with version 3.1 of Zuker's MFold program and the addition of low complexity regions determined using default settings of the SEG program31 of the Genetics Computer Group, Madison,Wisconsin. Note that the mRNA-synonymous strands of the displayed genes are all in the 'top' strand ('w-strand') of the 172 kilobase EBV genome, so that base numbering (abscissa) is in ascending order. |
|
In the DNA encoding the EBNA-1 repetitive element the PLI approaches 800 bases per kilobase so that there are relatively few pyrimidines to pair with purines for formation of the stems of DNA stem-loop secondary structures. Thus, the overall potential for forming secondary structure is greatly decreased as measured by the FONS value (Fig. 1c). The latter value decomposes into a base composition-dependent component (FORS-M; Fig. 1c) and a base order-dependent component (FORS-D; Fig. 1b)28. Compared with neighbouring regions, base order contributes poorly to the folding potential (assessed as negative kcal/mol) in the region of the repetitive element. Indeed, base order supplements base composition in constraining the folding potential (so most FORS-D values are slightly positive; Fig. 1b). Because secondary structure in DNA is likely to enhance recombination5,8, this predicts decreased recombination between markers corresponding to the N-terminal and C-terminal ends of the protein. Apart from the low complexity repetitive element in the EBNA-1 protein, there are other non-repetitive low complexity elements (Fig. 1a). Similar elements are encoded in neighbouring genes, but are at a lower abundance. Thus, the immediate upstream gene (1614 bases) encodes two small elements. The two immediate downstream genes (414 and 768 bases) each have only one small element. In many respects (discussed in Ref 27) the EBNA-1-encoding gene is different from all other EBV genes. Indeed, there are more similarities with genes of P. falciparum. |
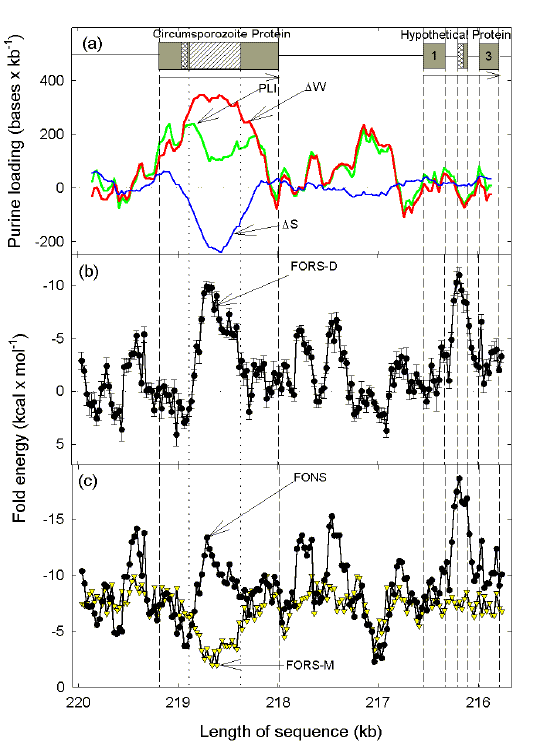
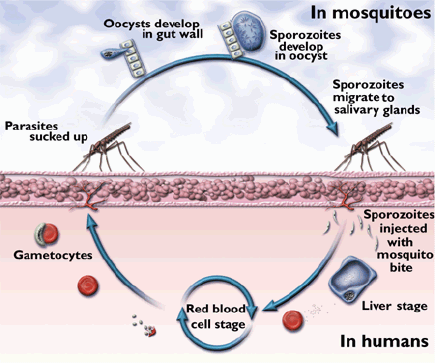
The circumsporozoite protein (CSP) of P. falciparum is expressed on sporozoites that are injected by infected mosquitoes and enter liver cells where there is the possibility of attack by cytotoxic T cells. The N-terminal and C-terminal segments correspond to polymorphic antigens that are evolving rapidly under positive selection3. There is a central low-complexity simple sequence element encoding octapeptide (NVDPNANP) or tetrapeptide (NANP) repeats, and one low-complexity, non-repetitive element (Fig. 2a). Downstream of the single exon CSP-encoding gene is a three exon gene encoding an unidentified 'hypothetical' protein. The upstream half of the small middle exon has a low complexity, non-repetitive, element. |
|
|
Fig. 2. Chargaff difference and fold analysis of the region of P. falciparum (clone 3D7) chromosome 3 encoding CSP and a hypothetical protein (GenBank: PFC0210c and PFC0205c). Vertical dotted lines indicate the boundaries of a low complexity, simple-sequence, repetitive element. Other details are as in Fig. 1. Note that the GenBank sequence was reversed so that the "bottom" strand ("c-strand") is the analysed strand (mRNA-synonymous strand) and genes formally designated as transcribing to the left are shown as transcribing to the right. Data are from a study by H. Y. Xue and D. R. Forsdyke (submitted for publication). |
|
The codons of the low complexity segments in the CSP gene tend to be A-loaded, and contribute to the generally high level of purine-loading (Fig. 2a). In regions corresponding to the N- and C-terminal segments this is achieved mainly by A bases, with a small contribution from G bases. For the middle segment this is achieved entirely by A bases, and ∆S is negative (i.e. there is an excess of C compared with G). The inclusion of the low-complexity regions can be considered as a device to enhance purine-loading, as in the case of the EBNA-1 antigen (Fig. 1). However, the fact that ∆S works against this suggests other factors are in play. Figures 2b and 2c show the potential of windows in the segment to form secondary structure. The base order-dependent component (FORS-D) is impaired in regions of the CSP gene corresponding to the N- and C-terminal segments of the protein (values tend to be positive); this is characteristic of sequences under positive selection34,41,44. Consistent with the high purine-loading of the repetitive middle segment, the base composition-dependent component of the folding potential (FORS-M) is greatly decreased in the middle segment (Fig. 2c); but this is compensated for by an increase in the base order-dependent component (Fig. 2b). Hence, the natural sequence shows a large increase in overall stem-loop potential (FONS) in this region (Fig. 2c). This suggests that, unlike the EBNA-1 encoding gene of EBV (Fig. 1), recombination might be enhanced between markers corresponding to the N- and C-terminal segments. Saul17 has speculated that the failure to find this to date is due to sample bias. In general, stem-loop potential in genes tends to be relegated to introns, especially in the case of genes under positive selection where introns may be more conserved than exons40,41,43. In the case of the CSP gene, what might have been an intron appears to have been retained and expressed in the protein sequence, probably because of an adaptive value at the RNA level (i.e. purine-loading) and/or at the protein level. Remarkably, while showing a relatively poor degree of purine-loading, the gene encoding the small hypothetical protein shows a similar stem-loop pattern, with a large central contribution of base order (Fig. 2b), but not base composition (Fig.2c), to the total folding energy (Fig 2c). This correlates with the insertion of two introns and a small exon much of which encodes a low complexity segment. |
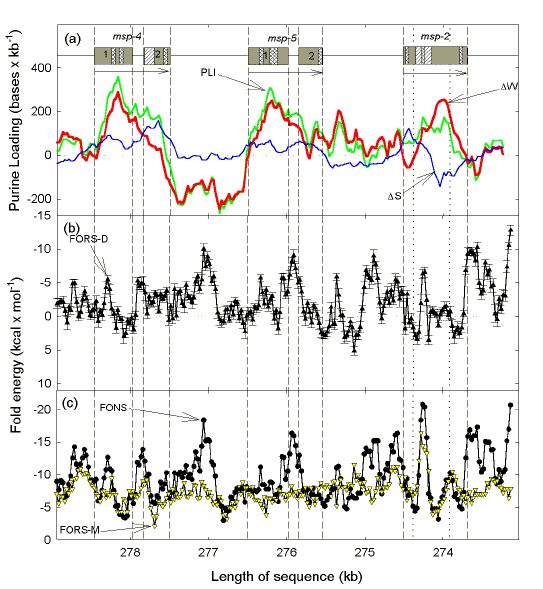
Merozoites in erythrocytes are not subject to T cell attack. Figure 3 shows a segment of chromosome 2 of P. falciparum with two double-exon genes encoding merozoite surface proteins 4 and 5 (MSP-4; MSP-5) that are of equal length, upstream of the gene encoding merozoite surface protein 2 (MSP-2)3. Two vertical dotted lines divide the single MSP-2 exon into conserved 5' and 3' segments, and a polymorphic immunodominant middle segment with a highly variable simple-sequence repeat in its 5'-half. |
|
|
Fig. 3. Chargaff difference and fold analysis of the region of P. falciparum chromosome 2 encoding MSP-4, MSP-5 and MSP-2 (GenBank: PFB310c, PFB305c, and PFB0300c). Two vertical dotted lines divide the MSP-2-encoding region into terminal conserved segments and a central, highly polymorphic, segment. Other details are as in Figs. 1 and 2. Data are from a study by H. Y. Xue and D. R. Forsdyke (2003; see below). |
|
Consistent with Szybalski's transcription direction rule5,37, all genes show purine-loading with A residues predominant, and intergenic regions tend to be negatively purine-loaded (i.e. pyrimidine-loaded; Fig. 3a). Purine-loading in the MSP-2-encoding gene is mainly contributed by A residues in the middle segment. Figures 3b and 3c show the potential of windows in the segment to form DNA secondary structure. Consistent with previous work40,41,43, folding energy tends to be lower (low negative, or positive, values) in exons, and greater (high negative values) in introns and intergenic regions. While the base composition-dependent component of the folding energy (FORS-M) remains relatively constant, the base order-dependent component (FORS-D) usually makes a major contribution to the total folding energy (FONS). Consistent with being under positive selection3, the FORS-D component of the folding energy is considerably impaired (values become positive) in parts of the gene encoding MSP-2, especially at the 5'-end of the middle segment and in the 3'-segment (Fig. 3b). In the region of the repeats both base order and base composition contribute to the high overall folding energy value (FONS). Thus, this repeat region appears to be an adaptation for enhancing the potential for DNA secondary structure, hence facilitating recombination. In this respect, the region resembles an intron. |
|
Selective pressures conserving bases that, while not determining amino acids, determine GC%, purine-loading and fold potential, should be sufficient to account for low rates of synonymous mutations in P. falciparum. Thus, it may be incorrect to rest on such low rates the argument that existing species members are derived from a relatively recent ancestor. Distinctive features of its genome (low GC%, many low complexity segments) may be leading us to understand that the herald of the demise of so many human beings is now heralding that of the neutral theory and of the assumptions that rest upon it. Acknowledgements I thank Michael Zuker for his nucleic acid folding programs and Jim Gerlach for assistance with computer configuration. Queen's University supports my bioinformatics web-pages. |
References
|
1 Schaap, T. (1971) Dual information in DNA and the evolution of the genetic code. J. Theor. Biol. 32, 293-298 2 Kimura, M. (1977) Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 267, 275-276 3 Hughes, A.L. (1999) Adaptive Evolution of Genes and Genomes. Oxford University Press 4 Bernardi, G. (2001) Misunderstandings about isochores. Gene 276, 3-13 5 Forsdyke, D.R. and Mortimer, J.R. (2000) Chargaff's legacy. Gene 261, 127-137 6 Forsdyke, D.R. (2001a) Adaptive value of polymorphism in intracellular self/not-self discrimination. J. Theor. Biol. 210, 425-434 7 Forsdyke, D.R. (2001b) Functional constraint and molecular evolution. Encyclopedia of Life Sciences (Vol. 7), pp. 396-403, Nature Publishing Group 8 Forsdyke, D.R. (2001c) The Origin of Species, Revisited, McGill-Queen's University Press 9 Rich, S.M. et al. (1998) Malaria's Eve: evidence of a recent population bottleneck throughout the world populations of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 95, 4425-4430 10 Rich, S.M. and Ayala, F.J. (1998) The recent origin of allelic variation in antigenic determinants of Plasmodium falciparum. Genetics 150, 515-517 11 Rich, S.M. and Ayala, F.J. (1999) Reply. Parasitol. Today 15, 39-40 12 Rich, S.M. et al. (2000) The origin of antigenic diversity in Plasmodium falciparum. Parasitol. Today 16, 390-396 13 Rich, S.M. and Ayala, F.J. (2000) Population structure and recent evolution of Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 97, 6994-7001 14 Arnot, D.E. (1989) Malaria and the major histocompatibility complex. Parasitol. Today 5, 138-142 15 Arnot, D.E. (1991) Possible mechanisms for the maintenance of polymorphisms in Plasmodium populations. Acta Leidensia 60, 29-35 16 Saul, A. and Battistuta, D. (1988) Codon usage in Plasmodium falciparum. Mol. Biochem. Parasitol. 27, 35-42 17 Saul, A. (1999) Circumsporozoite polymorphisms, silent mutations and the evolution of Plasmodium falciparum. Parasitol. Today 15, 38-39 18 Hughes, A.L. and Verra, F. (1998) Ancient polymorphism and the hypothesis of a recent bottleneck in the malaria parasite Plasmodium falciparum. Genetics 150, 511-513 19 Hughes, A.L. and Verra, F. (2001) Very large long-term effective population size in the virulent human malaria parasite Plasmodium falciparum. Proc. R. Soc. London. Ser. B. 268, 1855-1860 20 Hughes, A.L. and Verra, F. (2002) Extensive polymorphism and ancient origin of the human malaria parasite. Trends Parasitol 18, 348-355 21 Volkman, S.K. et al. (2001) Recent origin of Plasmodium falciparum from a single progenitor. Science 293, 482-484 22 Hartl, D. L. et al. (2002) The paradoxical population genetics of Plasmodium falciparum. Trends Parasitol. 18, 266-273 23 Grantham, R. (1980) Workings of the genetic code. Trends Biochem. Sci. 5, 327-331 24 Bernardi, G. and Bernardi, G. (1986) Compositional constraints and genome evolution. J. Mol. Evol. 24, 1-11 25 Knight, R.D. et al. (2001) A simple model based on mutation and selection explains trends in codon and amino acid usage and GC composition within and across genomes. Genome Biol. 2, research0016.1-0016.8 26 Lao, P.J. and Forsdyke, D.R. (2000) Thermophilic bacteria strictly obey Szybalski's transcription direction rule and politely purine-load RNAs with both adenine and guanine. Genome Res. 10, 228-236 27 Cristillo, A.D. et al. (2001) Double-stranded RNA as a not-self alarm signal. J. Theor. Biol. 208, 475-491 28 Forsdyke, D.R. (1998) An alternative way of thinking about stem-loops in DNA. J. Theor. Biol. 192, 489-504 29 Sueoka, N. (1961) Compositional correlation between deoxyribonucleic acid and protein. Cold. Spring Harb. Symp. Quant. Biol. 26, 35-43 30 Sharp, P.M. and Li, W.-H. (1987) The rate of synonymous substitution in eubacterial genes is inversely related to codon usage bias. Mol. Biol. Evol. 4, 222-230 31 Wan, H. and Wootton, J.C. (2000) A global complexity measure for biological sequences. AT-rich and GC-rich genomes encode less complex proteins. Comput. Chem. 24, 71-94 32 Pizzi, E. and Frontali, C. (2001) Low-complexity regions in Plasmodium falciparum proteins. Genome Research 11, 218-229 33 Forsdyke, D.R. (1999) Two levels of information in DNA. J. Theor. Biol. 201, 47-61 34 Forsdyke, D.R. (1995c) Reciprocal relationship between stem-loop potential and substitution density in retroviral quasispecies under positive Darwinian selection. J. Mol. Evol. 41, 1022-1037 35 Gardner, M.J. et al. (1998) Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science 282, 1126-1132 36 Bowman, S. et al. (1999) the complete nucleotide sequence of chromosome 3 of Plasmodium falciparum. Nature 400, 532-538 37 Szybalski, W. et al. (1966) Pyrimidine clusters in the transcribing strands of DNA and their possible role in the initiation of RNA synthesis. Cold Spring Harbor Symp. Quant. Biol. 31, 123-127 38 Salser, W. (1970) Discussion. Cold Spring Harb. Symp. Quant. Biol. 35, 19 39 Ball, L.A. (1973) Secondary structure and coding potential of the coat protein gene of bacteriophage MS2. Nature New Biol. 242, 44-45 40 Forsdyke, D.R. (1995a) A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol. Biol. Evol. 12, 949-958 41 Forsdyke, D.R. (1995b) Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. Mol. Biol. Evol. 12, 1157-1165 42 Seffens, W. and Digby, D. (1999) mRNAs have greater negative folding free energies than shuffled or codon-choice randomized sequences. Nucleic Acids Res. 27, 1578-1584 43 Barrette, I.P. et al. (2001) Introns resolve the conflict between base order-dependent stem-loop potential and the encoding of RNA or protein: further evidence from overlapping genes. Gene 270, 181-189 44 Forsdyke, D.R. (1996) Stem-loop potential in MHC genes: a new way of evaluating positive Darwinian selection. Immunogenetics 43, 182-189 45 Nussinov, R. (1984) Doublet frequencies in evolutionarily distinct groups. Nucleic Acids Res. 12, 1749-1763 46 Bossi, L. and Roth, J.R. (1980) The influence of codon context on genetic code translation. Nature 286, 123-127 47 Akashi, G. (1994) Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. J. Mol. Evol. 136, 927-935 End of paper End-Note: In July 2002 Mu et al. reported 100,000 to 180,000 years to the most recent common ancestor, based on an analysis of single nucleotide polymorphisms from 204 genes of chromosome 3 (Nature 418, 323-326). |
|
|
|
|
|
|
|
|
![]()
H. Y. Xue and D. R. Forsdyke
Mol.
Biochem. Parasitol. (2003) 128, 21-32.
Click Here for Journal
With
copyright permission from Els evier
| Abbreviations: AC%, percentage A + C; AG%, percentage A + G; EBNA1, Epstein-Barr Nuclear Antigen 1; EBV, Epstein-Barr Virus; FONS, folding of natural sequence; FORS-D, folding of randomized sequence difference; FORS-M, folding of randomized sequence mean; GC%, percentage G + C; ORF, open reading frame. |
![]()
3.1. Gene and intron lengths are not correlated
3.2. Low-complexity correlates with base composition of first and second codon positions
3.3. Low complexity regions in EBV
3.4. Low-complexity segments countermand or support fold potential
4.1. Do low complexity segments replace introns?
4.2. Low complexity correlates with base composition of amino acid-determining codon positions
![]()
Abstract
Protein segments that contain few of the possible twenty amino acids, sometimes in tandem repeat arrays, are referred to as containing "simple" or "low complexity" sequence. Many P. falciparum proteins are longer than their homologs in other species by virtue of their content of such low complexity segments that have no known function; these are interspersed among segments of higher complexity to which function can often be ascribed. If there is low complexity at the protein level there is likely to be low complexity at the corresponding nucleic acid level (departure from equifrequency of the four bases). Thus, low complexity may have been selected primarily at the nucleic acid level and low complexity at the protein level may be secondary. In this case the amino acid composition of low complexity segments should be more reflective than that of high complexity segments on forces operating at the nucleic acid level, which include GC-pressure and AG-pressure. Consistent with this, for amino acid-determining first and second codon positions, open reading frames containing low complexity segments show increased contributions to downward GC-pressure (revealed as decreased percentage of G +C) and to upward AG-pressure (revealed as increased percentage A + G). When not countermanded by high contributions to
AG-pressure, low complexity segments can contribute to base order-dependent
fold potential; in this respect they resemble introns. Thus, in P.
falciparum low complexity segments appear as adaptations primarily serving
nucleic acid level functions. |
|
Many eukaryotic genes consist of segments that are expressed as a mature transcript (exons), and of segments that are removed from the initial transcript (introns). Among both types of segment are segments of low complexity with biases towards individual bases or groups of bases, and segments of high complexity in which the four bases tend to be equally present. Exons have a narrow size range, with a peak at about 100 bases, and the sum of intron lengths tends to increase proportionately as the sum of exon lengths increases [1-5]. Notable exceptions are genes of the malarial parasite, Plasmodium falciparum. Here, as exon length increases there is a proportionate increase in low complexity sequence within exons. This is evident at the protein level with segments biased towards individual amino acids or groups of amino acids (usually less conserved, hydrophilic, sequences associated with non-globular protein domains) being dispersed among segments in which the 20 amino acids tend to be more evenly present (usually corresponding to conserved, globular, domains of defined function). Thus, Pizzi and Frontali noted that proteins of P. falciparum are often longer than their homologs in other species that are composed mainly of domains of defined function [6].
Since
low complexity at the nucleic acid level correlates with low complexity at the
protein level (e.g. poly(dT) encodes
polyphenylalanine), it is possible that a
primary selective influence operating at one level is the cause of low
complexity at both levels. Indeed, Wan and Wootton [7] noted for bacteria,
archaea, and eukaryotes that GC-rich genomes and GC-poor (AT-rich) genomes
have more low complexity segments in their proteins than genomes in which the
quantity of G + C is approximately equal to the quantity of A + T. Thus, it is
likely that "GC-pressure" (pressure to depart from equifrequency of these
base combinations) is a primary cause of a bias in amino acid composition of
proteins towards certain amino acids, which would tending to generate low
complexity segments in proteins. It is intuitively evident, and follows from
information theory (see
Methods), that sequences without base equifrequency
(e.g. TTTTTTTT) have less potential to transmit information than sequences
with base equifrequency (e.g.
ACGGCTTA). Thus, assuming a need to optimize
information-carrying capacity (i.e. there is not a redundancy of
information-carrying capacity), deviations from base equifrequency due to one
pressure should constrain the ability of a sequence to respond to other
pressures. Genomes under extreme GC-pressure, such as that of P. falciparum,
should be particularly informative in this respect. GC-pressure is but one of a variety of pressures appearing to operate directly on nucleic acid sequences, and is a major contributor to what has been referred to as the "genome phenotype" [10-12]. Among these pressures, "AG-pressure" (purine-loading pressure; [13]) can account for some prominent low complexity elements in herpesvirus proteins [14]. Evidence that this pressure might also generate low complexity segments in proteins of P. falciparum came from the observation that amino acid biases in low complexity segments do not correlate with properties of individual amino acids (hydrophobicity, volume, flexibility), but do correlate with the A content of their codons [6]. We here further examine the role of low complexity segments in the AT-rich P. falciparum genome where the third codon position, least related to amino acid composition, tends to be dominated by GC-pressure. The results assist our understanding of the roles of low complexity segments and of the introns that they appear to replace. Some biological implications of this work are reviewed elsewhere [15; see above paper]. |
|
P. falciparum sequences of chromosome 2 (AE001362; 209 predicted ORFs [16]) and of chromosome 3 (NC_000521; 215 predicted ORFs [17]), and an Epstein-Barr virus (EBV) sequence (V01555; 84 predicted ORFs [18]), were obtained from GenBank (National Center for Biotechnological Information, Bethesda, MD). A few genes that were difficult to evaluate (e.g. non-canonical start codons) were eliminated generating totals of 205 (chromosome 2), 206 (chromosome 3), and 65 (EBV). For detailed studies of individual genes, updated sequences were obtained from The Institute for Genomic Research or from the Sanger Centre.
2.2.
Low complexity segments
If the four bases of a nucleic acid sequence were distributed randomly, but with equal probability, along a sequence (i.e. at a particular position the uncertainty as to which base was coming next was maximum), then, by definition, each base would add 2 binary digits (bits) of information (4 = 22). If there were only one base, there would be no uncertainty as to which base was coming next, and each base would add 0 bits of information (1 = 20). Thus variation in nucleic acid sequences can be assessed in terms of their information content on a scale from 0 to 2. Similar considerations apply to protein sequences, where the scale is from 0 (1 = 20) to 4.322 (20 = 24.322). The quality measured can be referred to as "uncertainty," "entropy," or "complexity" [19]. This also relates to "compressibility," since a simple sequence is easier to characterize in terms that allow a decrease in total symbol number (e.g. "AAAAAA" can be written "6A"). By defining appropriate window sizes and cut-off points, sequences can be partitioned into low and high complexity segments. Thus, the SEG algorithm [7] as adapted for the GCG software suite (Accelrys Inc., San Diego, CA), was employed for determining low complexity segments in proteins using recommended default settings (window, 12 bases; trigger segment complexity 2.2 bits/residue; extension segment complexity 2.5 bits/residue). These settings are more stringent than those employed by Pizzi and Frontali [6] and no ORFs scored as 100% low complexity. Programs for Microsoft Excel were written in Microsoft Visual Basic to calculate base compositions (GC%, AG%, AC%) from species codon usage tables [20]. From four bases there are six combinations of two different bases, consisting of three mutually-exclusive pairs (GC, AT; AG, CT; AC, GT). Thus, implicit in values of GC% for a DNA segment are values for the reciprocating base pair, AT%. When AG% is 50% there are equal quantities of purines and pyrimidines (R and Y, respectively), and a parameter employed in previous work, the "purine-loading index," is then zero (reviewed in [10]). Base counts in windows of fixed size were carried out with the GCG program WINDOWS. Although window sizes of around 1000 nt were found optimal in previous work with various species [21], satisfactory results were obtained in the present work with a window size of 400 nt, which is close to that used for fold potential (see below). However, although moved along sequences in 25 nt steps, this window size does not give good resolution when exons and introns are small (e.g. first four exons of Fig. 6). "Pressure" is a useful intuitive concept implying some force or tendency with a directional component. As indicated in the text, the terms "GC-pressure" and "AG-pressure" are used here to indicate evolutionary forces for departures from base equifrequencies. However, currently there is no strictly defined usage in the literature. Thus, in some contexts, the effect of "GC-pressure" (i.e. a change in GC%) is considered to increase over the range 0% GC to 100% GC, while the effect of "AT-pressure" is considered to decrease correspondingly from 100% AT to 0% AT. Here we avoid the latter term, which is implicit in the former. Thus, in the present context the effect of "GC-pressure" is considered to increase both from around 50% GC to 100% GC ("upward GC-pressure") and from around 50% GC to 0% GC ("downward GC-pressure").
2.4.
Base Order-Dependent Fold Potential
Potential for single-stranded DNA at 37oC to form higher ordered structure at physiological salt concentrations (150 mM Na+ and 5 mM K+), was evaluated using version 3.1 of MFold [22]. Values were calculated for 200 nt windows moving along sequences in steps of 25 nt. Each value for the total folding energy ("folding of natural sequence" = FONS) was decomposed into a base composition-dependent component ("folding of randomized sequence mean" = FORS-M), and a base order-dependent component ("folding of randomized sequence difference" = FORS-D), as described previously [23-27]. |
|
|
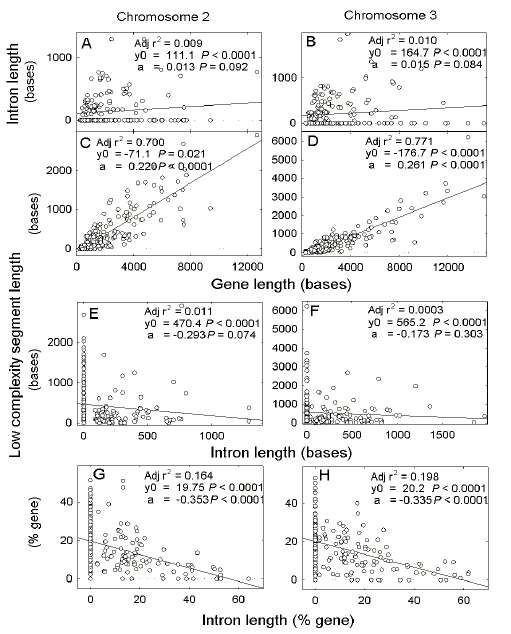
| Fig. 1.
Chromosomes 2
and 3 of P. falciparum. Relationship of lengths of introns (a, b) and
low complexity segments (c,d) to the lengths of the corresponding genes, and
relationship of either absolute (e, f) or percentage (g, h) lengths of low
complexity segments to the lengths of introns in the same genes.
Parameters of first order linear regression plots are shown in each figure: Adj r2, square of the adjusted correlation coefficient; y0, intercept at the ordinate; a, slope; P, probability that the preceding value is not significantly different from zero. |
|
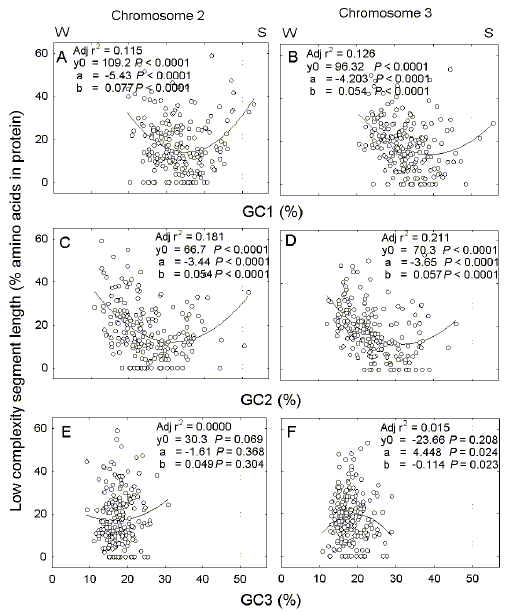
3.1. Gene and intron lengths are not correlated Unlike many eukaryotic genomes, there is no correlation between gene length and total intron length in chromosomes 2 and 3 of the P. falciparum genome (Figs. 1a, b). However, as noted by Pizzi and Frontali [6], many P. falciparum genes have low complexity segments and there is a close correlation between gene length and the combined lengths of these segments in a gene. Linear regression plots cross the abscissa at 323 bases (chromosome 2; Fig. 1c) and 680 bases (chromosome 3; Fig. 1d), indicating a gene length threshold below which low complexity segments do not occur. The low complexity segments seem to replace introns. However, introns and low complexity segments are not reciprocally interchangeable in that, as absolute intron length increases, there is no significant decline in length of low complexity segments (Figs. 1e, f). Thus, the relationship appears additive rather than reciprocal. It is only when the lengths are expressed as a proportion of gene length that a reciprocal relationship emerges (Figs. 1g, h). |
|
|
|
Fig. 2.
Variation of low
complexity segment length in proteins encoded by chromosomes 2 (a,c,e) and 3
(b, d, f) of P. falciparum with the GC-content of first (a, b), second
(c, d) and third (e, f) codon positions.
Each point represents an individual ORF. Parameters of second order linear regression plots are shown in each figure. W and S indicate the enrichment for either the W bases (A + T) at low GC%, or the S bases (G + C) at high GC%. Details are as in Figure 1. Calculated minima correspond to GC% values of (a) 35.3, (b) 38.9, (c) 31.8, (d) 32.0, (e) 16.4, and (f) 19.5. |
|
3.2.
Low-complexity correlates with base composition of first and second codon
positions For each gene of chromosomes 2 and 3, the percentage content of low complexity segment was plotted against percentage G+C (GC%) for each of the three codon positions in the ORF. Essentially the same results were obtained with both chromosomes (Fig. 2). Average GC% values become progressively less as codon position changes from first position (Figs. 2a,b) to third position (Figs. 2e,f) [28]. The extreme value of the third position (18 %) indicates that it has the most flexibility to respond to downward GC-pressure. This is largely independent of the percentage of low complexity segment in ORFs (low adjusted r2 values in Figs. 2e, f). Thus, whatever their content of low complexity sequence, on the basis of their third codon position GC percentages ORFs contribute to downward GC-pressure. On the other hand, data points for first and second codon positions fit to second order linear regressions as quadratic parabolas (y = y0 + ax + bx2) with minima corresponding to ORFs with low percentages of low complexity segment (Figs. 2a-d). Most data points relate to the leftward limbs of the parabolas (especially in the case of second codon positions; Figs. 2c,d). For first and second codon positions, the parabolic relationships show that the acquisition by certain ORFs of GC percentages lower than the GC percentages corresponding to minimum low complexity content, correlates with an increased content of low complexity segment. Thus, the low complexity segments could be evolutionary adaptations allowing ORFs, through the compositions of first and second codon positions, to contribute to extreme base compositional demands (i.e. downward GC-pressure) that could not be met by high complexity segments alone. |
|
|
|
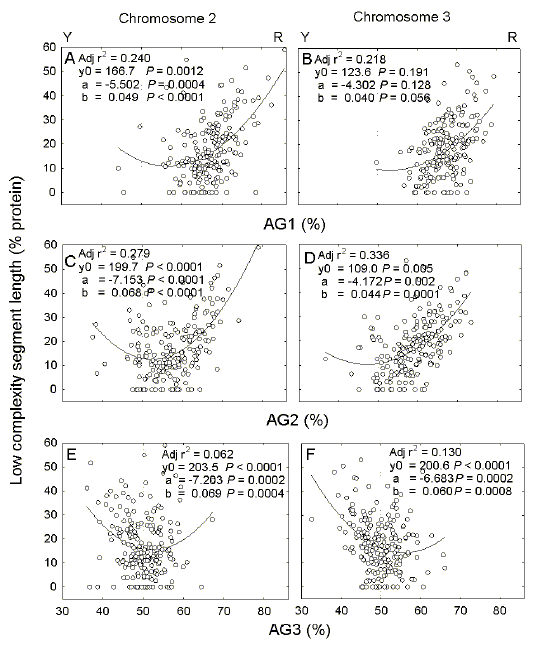
Fig. 3.
Variation of low
complexity segment length in proteins encoded by chromosomes 2 (a,c,e) and 3
(b, d, f) of P. falciparum with the AG-content of first (a, b), second
(c, d) and third (e, f) codon positions.
Y and R indicate the enrichment for either the Y bases (C + T) at low AG%, or the R bases (A + G) at high AG%. Details are as in Figure 2. Calculated minima correspond to AG% values of (a) 56.1, (b) 53.8, (c) 52.6, (d) 47.4, (e) 52.2, and (f) 55.7. |
|
Similar results were obtained when AG-pressure was evaluated. Departure from equifrequency of bases (50% purine: 50% pyrimidine) is greatest in the case of first codon positions. Consistent with the "RNY rule" (purine for first codon base, pyrimidine for third codon base; [29]), average AG% values become progressively less as codon position changes from first position (Figs. 3a, b) to third position (Figs. 3e, f). Values for first and second codon positions can best be fitted to second order linear regressions; most data points relate to rightward limbs of parabolas (Figs. 3a-d), indicating a positive correlation between purine content (i.e. upward AG-pressure) and ORF content of low complexity segment. Values for third codon positions show poor correlations (adjusted r2 values of 0.062 and 0.130; Figs. 3e, f) indicating that distinction between ORFs with low and high percentages of low complexity segment length is least for this position. |
|
|
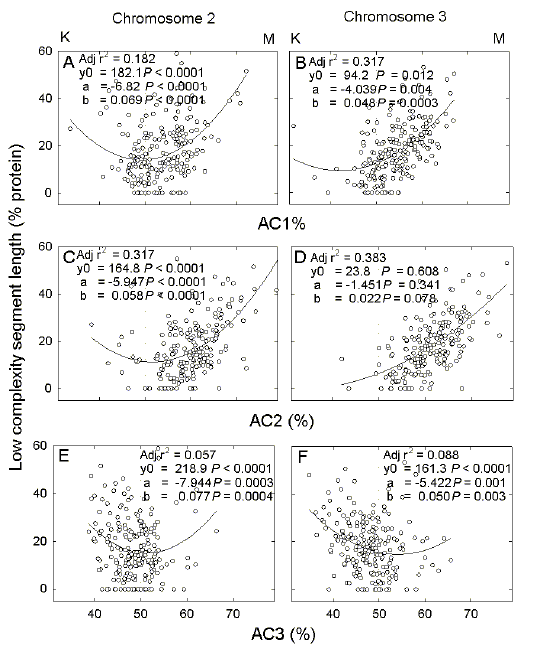
| Fig. 4. Variation of low complexity segment length in proteins encoded by chromosomes 2 (a,c,e) and 3 (b, d, f) of P. falciparum with the AC-content of first (a, b), second (c, d) and third (e, f) codon positions. K and M indicate the enrichment for either the K bases (G + T) at low AC%, or the M bases (A + C) at high AC%. Details are as in Figure 2. |
| For
symmetry, the hypothetical entity "AC-pressure" was also evaluated. The results were
similar to those for GC-pressure and AG-pressure with best regression
correlations in the case of first and second codon positions (Fig.
4).
3.3.
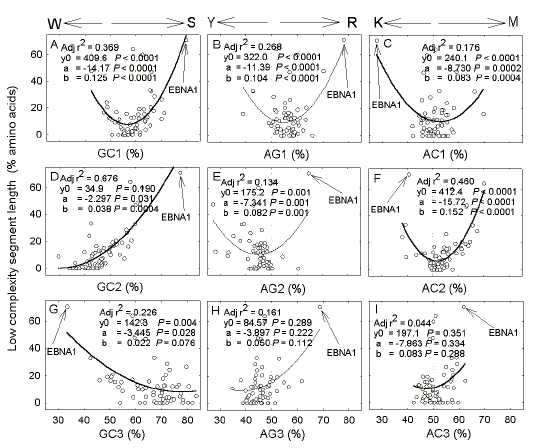
Low complexity regions in EBV In contrast to P. falciparum (low genome GC%; high ORF AG%) the more compact EBV genome is 60% GC and has low AG% in ORFs (i.e. pyrimidine-loading). An exception to the latter is the gene encoding Epstein-Barr Nuclear Antigen 1 (EBNA1), which is highly purine-loaded, mainly by virtue of a long low complexity segment [14,15]. In general, ORFs of EBV encode less low complexity amino acid sequences than those of P. falciparum. Thus, on average, ORFs of EBV are 13.3%"1.9% low complexity, whereas the percentages in P. falciparum are 17.6%"0.9% (chromosome 2) and 18.1%"0.8% (chromosome 3). |
|
|
| Fig. 5. Variation of low complexity segment length in EBV proteins with GC-content (a, d, g), AG-content (b, e, h), and AC-content (c, f, i) of first (a, b, c), second (d, e, f) and third (g, h, i) codon positions. The point corresponding to the ORF encoding EBNA1 protein is indicated with an arrow. Details are as in Figures 2-4. |
| Like P. falciparum, in EBV low complexity percentage values can be fitted to quadratic parabolas, with most values being located near minima (Fig. 5). In all plots the extreme low complexity value corresponding to the EBNA1-encoding gene lies at one extreme of the curve. The first and second codon positions tend to be the best predictors of low complexity segment length, particularly in the case of GC% (adjusted r2 = 0.67 for second codon positions; Fig. 5d). |
|
3.4.
Low-complexity segments countermand or support fold potential The long low complexity sequence in the EBNA1 gene appears to countermand the potential of the encoding DNA duplex to extrude single-stranded DNA that adopts stable stem-loop secondary structure [14,15]. Similar studies were carried out on P. falciparum genes, and we show here results from a multiexon gene (Fig.6), a two exon gene (Fig. 7) and a single exon gene (Fig. 8). |
|
|
|
Fig. 6. Variation of AG content
(a) and fold energy (b, c) for sequence windows of 400 nt (a) or 200 nt (b, c)
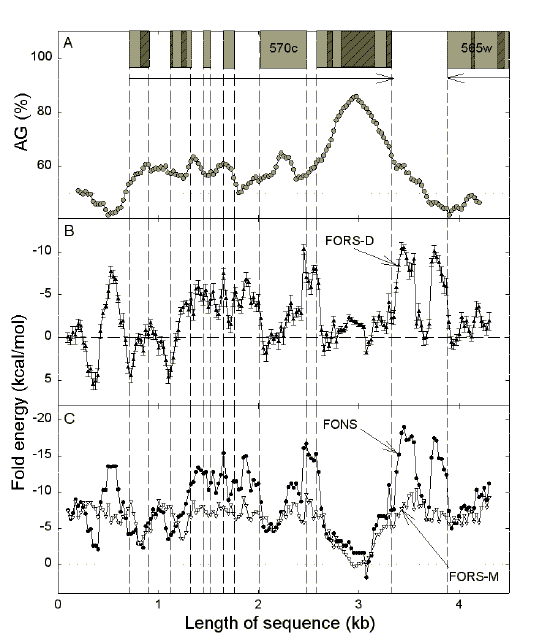
in the region containing PFC0570c. This is an AG-rich, multi-exon, gene of chromosome 3 of P. falciparum that encodes a hypothetical protein of unknown function. Each total folding energy value (FONS; filled circles in (c)), is decomposed into a base composition-dependent component (FORS-M; open triangles in (c)), and a base order-dependent component (FORS-D; closed triangles in (b) with standard errors of the mean). Exons are represented as grey boxes with the direction of transcription indicated by the horizontal arrow. Low complexity segments within exons are indicated as dark grey boxes with slashed lines. Vertical dashed lines indicate exon borders. The gene is formally designated as transcribing to the left (hence the "c," not "w," in the gene name), so the "bottom" strand of the GenBank sequence is shown here. Thus, for PFC0570c this is the mRNA-synonymous strand, whereas for the neighbouring gene transcribed in the opposite direction (PFC0565w) this is the mRNA-template strand. [It is of interest to note that the original GenBank record (NC_000521) assigned only 3 exons to PFC0570c, which correspond to the present exons 5 and 6 plus a small 45 base first exon 155 bases upstream. The version used here is an update from the Sanger Centre (Release 63, version 12; April 2000; contig MAL3P4)]. |
| Figure 6a shows a gene from chromosome 3 (PFC0570c) with a large low complexity segment in the 6th exon. This corresponds to a region of high AG%, consistent with a purine-loading role for the low complexity segment. In keeping with Szybalski's transcription direction rule (high top-strand purine content when transcription is to the right [10]), AG values are greater than 50% in exons, and decrease in introns and intergenic DNA. Conversely, as noted previously [23,24], exons (especially large exons) tend to have low total fold potential, whereas introns (here introns 2-5) and flanking intergenic DNA tend to have high fold potential. Base order contributes more to fold potential (Fig. 6b) than base composition (see values for FORS-M in Fig. 6c), which tends to remain constant. |
|
|
|
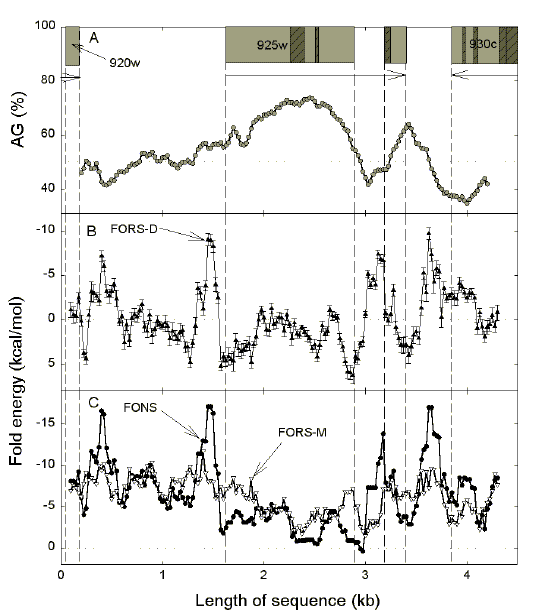
Fig. 7. Variation of AG-content (a) and fold energy (b, c) for
sequence windows in the region containing PFC0925w, a very AG-rich, two
exon gene of chromosome 3 of P. falciparum that encodes a hypothetical
protein of unknown function. The sequence shown is that of the "top" strand which is the mRNA-synonymous strand for PFC0925w. Details are as in Figure 6. [It is of interest to note that the original GenBank record (NC_000521) assigned only one exon to PFC0925w. The version shown here is an update from the Sanger Centre (Release 63, version 7; May 2000; contig MAL3P7)]. |
| Figure 7 shows a gene with only one intron (PFC0925w). Here again exons tend to be AG-rich, and in exon 1 this is prominent in the region of low complexity segments. There is a low complexity segment at the 5' end of exon 2 where AG-pressure is low. However the neighbouring intron segment has high base order-dependent fold potential; by virtue of the 0.2 kb window size, this would be partly contributed to by the exonic low complexity segment. As noted previously for neighbouring ORFs transcribed in different directions [30], with PFC0925w and PFC0930c there is a switch at the interface from purine-loading (AG > 50%) to pyrimidine-loading (AG < 50%); thus, whatever the transcription direction, mRNA-synonymous strands are always purine-loaded. This can also be discerned in the case of PFC0570c and PFC0565w (Fig. 6). |
|
|
|
Fig.
8.
Variation of AG-content (a) and fold energy (b, c) for sequence windows
in the region containing the single exon gene encoding liver stage antigen 1
(LSA1; GenBank accession X56203).
Details are as in Figure 6, except that there is a long region containing a 17 amino acid repeat (white box with slashed lines), which was not recognized as low complexity with the default settings of SEG; the simple sequence repeat emerged on elevating the trigger segment complexity threshold from 2.2 to 2.4 bits/residue. Immunogenic epitopes T1, LSA-J, Is6, T3 and T5 are shown as open white boxes.Vertical dashed lines indicate exon borders. Vertical dotted lines indicate the borders of the 17 amino acid repeat region. The average value for base order-dependent fold potential (FORS-D) in the repeat region is -4.80 se 0.25 kcal/mol, which is significantly different from average values for the region 5' of the repeat region (0.70 se 0.05; P = 0.0005) and for the region 3' of the repeat region (-2.62 se 0.46; P = 0.0005). |
|
The genes shown in Figures 6 and 7 have so far no assigned function; our data support a revised exon-intron assignment (see legends to figures), which has replaced that originally proposed [17]. Figure 8 shows a similar analysis of the long single exon gene encoding liver stage antigen 1 (LSA1), a major target for malaria immunotherapy [31]. Here there is a long region containing a 17 amino acid repeat (white box with slashed lines) that required a slight adjustment of the SEG program settings (see figure legend) for recognition as low complexity. Immunogenic epitopes T1, LSA-J, Is6, T3 and T5 correspond mostly to the non-repeat 5' and 3' regions, and tend to be separate from low complexity segments. In this respect the low complexity segments, as expected, do not function at the protein level. The entire region encoding the 17 amino acid repeat makes a major contribution both to purine-loading (AG%; Fig 8a), and to fold potential (Figs. 8b, c). The latter is largely by virtue of the contribution to base order-dependent fold potential (Fig. 8b). Similar analyses were carried out on several other genes of various functional classes, with results consistent with those shown here (e.g. circumsporozoite protein and merozoite surface proteins, 2, 4 and 5; see Ref. [15]). |
4.1. Do low complexity segments replace introns?This question is prompted by the observation that, whereas increase in total low complexity segment length is directly related to gene length (Figs. 1c,d), in P. falciparum this does not apply to intron length (Figs. 1a,b). The present work shows that quantitatively (with respect to absolute lengths) introns and low complexity segments are not mutually exchangeable (Figs. 1e, f). Furthermore, the low complexity segments tend to be of high AG% (purine-loading of ORFs), whereas introns tend to be of low AG% (i.e. tending towards pyrimidine-loading [34,35]). The question might be further addressed by comparing intron locations in regular eukaryotic genes (small exons and multiple introns), with the positions of low complexity segments in homologous genes of P. falciparum that have no, or few, introns. However, intron locations often show no relationship to defined protein functional domains [ 32,33], whereas low complexity segments predominate between defined protein functional domains (i.e. proteins with these superfluous segments are longer than their homologues [6]). Introns appear to facilitate base order-dependent fold potential, a function likely to be of importance for recombination [23]. This is particularly evident in genes under positive Darwinian selection, and with overlapping genes [24,25,36]. When contributing to AG-pressure (i.e. deviations from equifrequencies of A + G and C + T), low complexity segments can countermand fold potential (Figs. 6, 7) [14,15]. This is because the stems in stem-loop structures should be favoured optimally when AG is at 50% (i.e. when there are likely to be equal quantities of the Watson-Crick pairing bases [26]). Thus, the combined demands of purine-loading and protein-encoding may have been so severe as to instigate the generation of sequence segments that do not make these demands (i.e. the generation of introns). Here the potential to form secondary structure would be less constrained [11,12,23].
However,
when the contribution to AG-pressure is not too high, a low complexity segment
can support fold potential by appropriately ordering bases (base
order-dependent stem-loop potential; Fig. 8b). This also applies in the case
of the single chromosome 3 ORF encoding the P. falciparum
circumsporozoite protein which, like the LSA1-encoding gene, has a long low
complexity segment which contributes both to AG-pressure and to base
order-dependent fold potential (see Ref.
[15]). Furthermore, of three
merozoite surface protein ORFs tandemly arranged on chromosome 2 (MSP2,
MSP4, MSP5, which may have arisen from the replication of a
single original gene), two have introns which accommodate increased base
order-dependent fold potential; the third (MSP-2) has no
intron, but
appears to accommodate the potential by virtue of a low complexity segment
that also makes a small contribution to AG-pressure (see Ref.
[15] on this page above). We have evaluated the relationship between low complexity and the frequencies of various pairs of bases, - GC%, AG% and AC%. Of these GC% and AG% have recognized (albeit sometimes controversial) roles at the nucleic acid level (reviewed in Ref. [10]). AC% completes the triad, but is currently an abstract entity of uncertain significance. Since low complexity is defined as a departure from equifrequencies of bases, it is not surprising that proteins with high percentages of low complexity segments correspond to the extremes of base pair percentages (Figs. 2-4). The issues are whether low complexity results from selective forces, and if so whether these forces operate primarily at the nucleic acid level or at the protein level. Low complexity minima would be expected to be close to the 50% values for base compositions. However, a segment which is 50% for A + G may not also be 50% for G + C, so precise correspondence is unlikely. The minima in plots of complexity against percentage base compositions (Figs. 2-5) may depart from 50% for this reason, and also because of pressures on individual codon positions. For example, tRNA anticodons are flanked by invariant 5' pyrimidine and 3' purines which participate in the stacking interactions involved in codon-anticodon recognition [37,38]. This can predispose first codon positions to be purine-rich and third codon positions to be pyrimidine-rich [29]. Thus, a preference in P. falciparum for asparagines (codons AAY) over lysine (codons AAR) would not necessarily imply a protein level adaptation, as has been suggested [6]. A recent comparison between the genomes of Plasmodium vivax and Plasmodium falciparum also indicates primary selective forces acting at the nucleic acid level [39]. Unlike the first and second codon positions, the base composition of third codon positions tends not to correlate with the percentage of low complexity segment in proteins, especially in the case of GC% (Figs. 2-4). This suggests that, for this position, low and high complexity segments are equally free to respond to base compositional pressures. First and second positions would be constrained by other pressures. In high complexity protein segments, mainly corresponding to conserved globular domains, these pressures would include pressures to retain specific protein function. On the other hand, the protein-encoding of low complexity segments would be largely determined by the base compositional pressures, and accordingly such segments would mainly correspond to non-globular hydrophilic surface domains which do not play a critical role in protein structure and function [6]. For example, the long low complexity element in EBNA1 protein encodes a glycine-alanine repeat that can be removed without impairing known EBNA1 functions [40].
We
conclude that many low complexity segments exist in proteins of P.
falciparum because of evolutionary forces acting at the nucleic acid
level. The nature of the functions served by these forces is discussed
elsewhere [10-12]. Any functions at the protein level, while not excluded
[41,42], would have been derived secondarily. The results obtained here with P.
falciparum (Figs.
2-4), are similar to those obtained with certain ORFs of
herpesvirus genomes (Fig. 5
[14,15]). Hence,
our conclusion may apply to low complexity segments in other genomes. Further
implications relating to the possibility of a recent P. falciparum
population bottleneck and the neutral theory [9] are discussed elsewhere
[15; see above]. Acknowledgements We thank Chris Madill for programs that display base compositions in the context of other sequence features, and James Mortimer for programs that extract base compositions from codon usage tables. Michael Zuker (Rensselaer Institute) kindly provided programs that fold DNA. Access to the GCG suite of programs was provided by the Canadian Bioinformatics Resource (Halifax, Nova Scotia). Jim Gerlach assisted with computer configuration. |
|
[1] Naora H, Deacon NJ. Relationship between the total size of exons and introns in protein-coding genes of higher eukaryotes. Proc. Nat. Acad. Sci. USA 1982;79: 6196-6200. [2] Blake C. Exons - present from the beginning? Nature 1983;306: 535-537. [3] Smith MW. Structure of vertebrate genes: a statistical analysis implicating selection. J. Mol. Evol. 1988;27: 45-55. [4] Traut TW. Do exons code for structural or functional units in proteins? Proc. Natl. Acad. Sci. USA 1988;85: 2944-2948. [5] Sakharkar M, Passetti F, Souza JE de, Long M, Souza SJ de. ExInt: an exon intron database. Nucleic Acids Res. 2002;30: 191-194. [6] Pizzi E, Frontali C. Low-complexity regions in Plasmodium falciparum proteins. Genome Research 2001;11: 218-229. [7] Wan H, Wootton JC. A global complexity measure for biological sequences. AT-rich and GC-rich genomes encode less complex proteins. Comput. Chem. 2000;24: 71-94. [8] Sueoka N. Compositional correlation between deoxyribonucleic acid and protein. Cold. Spring Harb. Symp. Quant. Biol.1961;26: 35-43. [9] Kimura M. 1977. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature 1977;267: 275-276. [10] Forsdyke DR, Mortimer JR. 2000. Chargaff's legacy. Gene 2000;261: 127-137. [11] Forsdyke DR. Functional constraint and molecular evolution. In: Encyclopedia of Life Sciences. London: Nature Publishing Group, 2001;7: 396-403. [12] Forsdyke DR. The Origin of Species, Revisited. McGill: McGill-Queen's University Press, 2001. [13] Lao PJ, Forsdyke DR. Thermophilic bacteria strictly obey Szybalski's transcription direction rule and politely purine-load RNAs with both adenine and guanine. Genome Res. 2000;10: 228-236. [14] Cristillo AD, Mortimer JR, Barrette IH, Lillicrap TP, Forsdyke DR. Double-stranded RNA as a not-self alarm signal: to evade, most viruses purine-load their RNAs, but some (HTLV-1, Epstein-Barr) pyrimidine-load. J. Theor. Biol. 2001;208: 475-491. [15] Forsdyke DR. Selective pressures that decrease synonymous mutations in Plasmodium falciparum. Trends Parasitol. 2002;18: 411-418. [16] Gardner MJ, Tettelin H, Carucci DJ, et al. Chromosome 2 sequence of the human malaria parasite Plasmodium falciparum. Science 1998;282: 1126-1132. [17] Bowman S, Lawson D, Basham D, et al. 1999. The complete nucleotide sequence of chromosome 3 of Plasmodium falciparum. Nature 1999;400: 532-538. [18] Baer R, Bankier AT, Biggin MD, et al. 1984. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 1984;310: 207-211. [19] Konopka A.K. Towards mapping functional domains in indiscriminantly sequenced nucleic acids: a computational approach. In: Sarma RH, Sarma MH, editors. Human Genome Initiative and DNA Recombination. New York: Adenine Press, 1990;1: 113-125. [20] Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from the international DNA sequence databases: status for the year 2000. Nucleic Acids Res.2000;28: 292. [21] Bell S, Forsdyke DR. Accounting units in DNA. J. Theor. Biol. 1999;197: 51-61. [22] Zuker M. Calculating nucleic acid secondary structure. Curr. Opin. Struct. Biol. 2000; 10: 303-310. [23] Forsdyke DR. A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol. Biol. Evol. 1995;12: 949-958. [24] Forsdyke DR. Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. Mol. Biol. Evol. 1995;12: 1157-1165. [25] Forsdyke DR. Reciprocal relationship between stem-loop potential and substitution density in retroviral quasispecies under positive Darwinian selection. J. Mol. Evol. 1995;41: 1022-1037. [26] Forsdyke DR. An alternative way of thinking about stem-loops in DNA. J. Theor. Biol. 1998;192: 489-504. [27] Seffens W, Digby D. mRNAs have greater negative folding free energies than shuffled or codon-choice randomized sequences. Nucleic Acids Res. 1999;27: 1578-1584. [28] Musto H, Caccio S, Rodriguez-Maseda H, Bernardi G. Compositional constraints on the extremely GC-poor genome of Plasmodium falciparum. Memoria do Instituto Oswaldo Cruz 1997;92: 835-841. [29] Eigen M, Schuster P. The hypercycle. A principle of natural self organization. Part C. The realistic hypercycle. Naturwissenschaften 1978;65: 341-369. [30] Bell S, Forsdyke DR. Deviations from Chargaff's second parity rule correlate with direction of transcription. J. Theor. Biol. 1999;197: 63-76. [31] Kurtis JD, Hollingdale MR, Luty AJ.F, Lanar DE, Krzych U, Duffy PE. 2001. Pre-erythocytic immunity to Plasmodium falciparum: the case for an LSA1 vaccine. Trends. Parasitol. 2001;17: 219-223. [32] Stoltzfus A, Spencer DF, Zuker M, Logsdon JM, Doolittle WF. Testing the exon theory of genes: the evidence from protein structure. Science 1994;265: 202-207. [33] Weber K, Kabsch W. Intron positions in actin genes seem unrelated to the secondary structure of the protein. EMBO. J. 1994;13: 1280-1286. [34] Saul A, Battistuta D. Codon usage in Plasmodium falciparum. Mol. Biochem. Parasitol. 1998;27: 35-42. [35] Mrazek J, Kypr J. Biased distribution of adenine and thymine in gene nucleotide sequences. J. Mol. Evol. 1994;39: 439-447. [36] Barrette IH, McKenna S, Taylor DR, Forsdyke DR. Introns resolve the conflict between base order-dependent stem-loop potential and the encoding of RNA or protein: further evidence from overlapping genes. Gene 2001:270: 181-189. [37] Bossi L, Roth JR. 1980. The influence of codon context on genetic code translation. Nature 1980;286: 123-127. [38] Simonson AB, Lake JA. The transorientation hypothesis for codon recognition during protein synthesis. Nature 2002;416: 281-284. [39] Tchavtchitch M, Fischer K, Huestis R. Saul A. The sequence of a 200 kb portion of a Plasmodium vivax chromosome reveals a high degree of conservation with chromosome 3. Mol. Biochem. Parasitol. 2001;118: 211-222. [40] Wu H, Kapoor P, Frappier L. Separation of the DNA replication, segregation, and transcriptional functions of Epstein-Barr nuclear antigen 1. J. Virol. 2002;76: 2480-2490. [41] Tellam J, Sherritt M, Thomson S, et al. Targeting of EBNA1 for rapid intracellular degradation overrides the inhibitory effects of the gly-ala repeat domain and restores CD8+ T cell recognition. J. Biol. Chem. 2001;276: 33353-33360. [42] Heessen S, Leonchiks A, Issaeva N, et al. Functional p53 chimeras containing the Epstein-Barr virus Gly-Ala repeat are protected from Mdm2- and HPV-E6-induced proteolysis. Proc. Natl. Acad. Sci. USA 2002;99: 1532-1537. |
|
|
|
|
|
|
|
|
![]()
Muralidharan et al (2011) from studies of a P. falciparum protein with an asparagine repeat sequence, conclude that "the asparagine repeat is dispensible for protein expression, stability and function. The data point to a genomic mechanism for repeat perpetuation rather than a positive cellular role." This supports the argument made in our above paper.
Muralidharan V, Oksman A, Iwamoto M, Wandless TJ, Goldberg DE (2011) Asparagine repeat function in Plasmodium falciparum protein assessed via a regulatable fluorescent affinity tag. Proceedings of the Natural Academy of Sciences USA 108, 4411-4416.
Tian et al (2011) demonstrate similar quadratic regressions as in the above paper, for 25 protist genomes including P. falciparum. They conclude that "Genome nucleotide content is a key factor controlling the abundance of SSRs [single sequence repeats] or microsatellites. Simulations confirm the simple expectation that sequences biased toward either AT or GC will generate more small random SSRs by chance. -- The relationship between AT content and SSR abundance (either frequency or density) is always U-shaped, with a minimum for relatively balanced genomes and rising toward both extremes" of base composition. Thus GC% "explains up to half the variance and the abundance and motif constitution of SSRs". Hearty & Golding (2011) report a detailed study of various independent isolates of P. falciparum.
Tian X, Strassmann JE, Queller DC (2011) Genome nucleotide composition shapes variation in simple sequence repeats. Molecular Biology & Evolution 28, 899-909.
Hearty W & Golding GB (2011) Increased polymorphism near low-complexity sequences across the genomes of Plasmodium falciparum isolates. Genome Biology & Evolution 3, 539-550.
End Note (Feb 2018) Intrinsically Disordered Regions
Much of the above was summarized in various editions of my textbook on evolutionary bioinformatics. Too late to be mentioned in the 3rd edition (2016) was an interesting review of intrinsically disordered regions of proteins (Pancsa & Tompa 2016). This began with the statement that "Numerous DNA- and RNA-level functions are embedded in protein-coding regions, which constrains their [i.e. the protein's] structure, function and evolution." Thus the above arguments are gaining wider recognition among protein biochemists.
Pancsa R, Tompa P (2016) Coding regions of intrinsic disorder accommodate parallel functions. Trends in Biochemical Sciences 41: 898-906.
Go to Amino acids as placeholders in malaria parasites Click Here
Return to Bioinformatics Index Click Here
Return to HomePage Click Here
![]()
Last edited on 11 November 2020 by Donald Forsdyke