Amino Acids as Placeholders
Base Composition Pressures on Protein
Length in Malaria Parasites and Prokaryotes
Jonathan H. Rayment and Donald R. Forsdyke
Applied Bioinformatics
(2005) 4, 117-130Submitted for publication 15 Nov 2004 . Accepted 1st March 2005
Copying of this article, including placing at another website, requires the written permission of Adis International Ltd for Walters Kluwer Health, the copyright owner and only authorized source. Please note that this version differs from the publisher's version with respect to use of coloured emphasis and insertion of numerical data within figures (which the publisher would not allow in the final version). In our opinion this early version is easier on the reader.
![]()
Length Effects Depend on Species Base Compositions
Length Effects Depend on Codon Positions
Incomplete Genomes Confirm Complete
Protein Length Constrained in Thermophiles
![]()
| Abstract:
The composition and sequence of amino acids in a protein may serve the underlying needs of the nucleic acids that encode that protein (the genome phenotype). In extreme form, amino acids become mere placeholders being inserted between functional segments or domains and, apart from increasing its length, playing no role in the specific function or structure of a protein (the conventional phenotype). We studied the genomes of two malarial parasites and of 521 prokaryotes (144 complete) that differ widely in GC% and optimum growth temperature. Malarial parasites show distinctive responses to base compositional pressures that increase as protein lengths increase. A low GC% species (P. falciparum) is likely to have more placeholder amino acids than an intermediate GC% species (P. vivax), so that homologous proteins are longer. In prokaryotes GC% is generally greater and AG% is generally less in open reading frames (ORFs) encoding long proteins. The increased GC% in long ORFs increases as species GC% increases, and decreases as species AG% increases. In low and medium GC% prokaryotic species, increases in ORF GC% as encoded proteins increase in length are largely accounted for by the base compositions of first and second (amino acid-determining) codon positions. In high GC% prokaryotic species first and third (non-amino acid-determining) codon positions play this role. Thus, in low and medium GC% prokaryotes placeholder amino acids are likely to be well defined, corresponding to codons enriched in G and/or C at first and second positions. In high GC% prokaryotes placeholder amino acids are likely to be less well defined. Increases in ORF GC% as encoded proteins increase in length are greater in mesophiles than in thermophiles, which are constrained from increasing protein lengths in response to base composition pressures. |
![]()
P roteins of the parasite responsible for the most severe form of malaria, Plasmodium falciparum, are much longer than their homologues in other species. Such differences in protein lengths between species might be explained as due to interspecies differences in associated functions, such as regulation. However, increased protein lengths in Plasmodium falciparum are due to the insertion of amino acids that appear to play no role in protein function. The amino acids act as mere "placeholders," serving the needs of the encoding nucleic acids for distinctive base compositions, not the needs of the encoded protein.[1-3] Thus, the composition and sequence of amino acids in a protein serve not only the functions of the protein as determined by classical Darwinian natural selection (the conventional phenotype), but also the functions of the corresponding nucleic acid (the genome phenotype).[4,5] Any conflicts between these functions [6,7] must be resolved to maximize genetic fitness, namely the number and fertility of descendents.Since the genetic code is degenerate, conflicts can often be resolved by appropriate choice of codons. Thus, pressure for a high genomic GC% can be resolved by the selective use of GC-rich codons. Failing this, amino acids with similar properties can be exchanged (e.g. A-rich codons for the basic amino acid lysine can be exchanged for GC-rich codons for the basic amino acid arginine). Failing this, amino acids with appropriate codons can be inserted into the protein, thus increasing protein length. Here the amino acids are primarily placeholders, their presence indicating that the bases in their codons may serve needs of the genome phenotype that cannot be satisfied by codon degeneracy or amino acid substitution. The presence of placeholder amino acids is suggested by gaps in protein alignments, which can correspond to low complexity, simple sequence elements.[1] In general, placeholder amino acids cannot exist in regions critical for protein function, such as the active site of enzymes. However, either singly or in groups, these extra amino acids can exist in less critical regions. In a protein with only one domain the less critical regions must be within that domain; this might affect the structure of the domain in a way that might compromise protein function. In multidomain proteins extra amino acids can exist between domains, often without compromising protein function. Since multidomain proteins tend to be longer than single domain proteins, longer proteins should more readily serve the needs of the genome phenotype, hence growing even longer due to interdomain insertions. Thus, long proteins might extend adaptations already evident in short proteins, or there might be adaptations specific for long proteins. A general comparison of short (putatively monodomain) protein-encoding sequences with long (putatively multidomain) protein-encoding sequences, should have the potential to provide information on evolutionary forces contributing to the genome phenotype. This should assist studies which assume that amino acids serve protein function. Such studies include attempts to understand and manipulate (e.g. drugs by design) protein function by the direct examination of amino acid sequences and their folding into higher ordered structures. Departures from base equifrequency constrain protein-encoding regions.[8] Thus, a need for interdomain insertions to satisfy pressures on the genome phenotype should be more evident in genomes with extreme average base composition values. Proteins encoded by the low GC% genome of Plasmodium falciparum are longer than their homologues in other species, due to the insertion of interdomain simple sequence elements.[1-3] Similarly, the major latency protein encoded by the high GC% genome of Epstein-Barr virus has a long interdomain insertion of a simple sequence element that may be removed without impairing known protein functions.[9] To what extent might this apply to genomes with less extreme departures from base equifrequency? Base compositional pressures being an important aspect of the genome phenotype, we here report relationships between base compositions and protein lengths in various genomes that range widely in their average GC% values. Since many prokaryotic genome sequences are available, after an initial comparison of the intermediate GC% genome of Plasmodium vivax with the low GC% genome of P. falciparum, whose interdomain insertions have been well characterized, this paper focuses on prokaryotes, including thermophiles where purine-loading (high AG%) is prevalent.[10-13]
|
| Data and Terminology
Data
Clearly, the selection of genomes for complete sequencing is limited and
not arbitrary. In the case of incompletely sequenced genomes there is a
further non-arbitrary selection of certain genes from within each
genome. However, at this time there are far more incompletely sequenced
genomes than completely sequenced genomes. So the advantages of studying
the fully sequenced genomes of a limited range of species have to be
weighed against the advantages of studying a much larger range of
species with partially sequenced genomes. Some general agreement has
been found between studies of a few completely sequenced genomes and
those of many partially sequenced genomes.[17] As part of the
present study, 377 incompletely sequenced prokaryotic genomes, for each
of which the sequences of 20 or more ORFs were available, were added to
the above 144 completely sequenced genomes in GenBank release 143, for a
total of 521 species (482 eubacteria and 39 archaebacteria). Values for
the optimum growth temperature of 293 of these species were found in the
Prokaryote Growth Temperature Database (PGTdb; http://pgtdb.csie.ncu.edu.tw). The term "base pressure" (e.g. "GC-pressure") acknowledges that some change in base composition can occur, but does not specify a cause.[18] Hypothetically, cell metabolism might produce an excess of a particular base (e.g. A) and this might result in a relative excess of that base in nucleic acids; or a base such as C might be unstable (e.g. due to deamination) and this might result in a relative lack of that base in nucleic acids; or enzymes for nucleic acid synthesis or repair might be intrinsically biased towards certain bases ("directional mutation pressure"); or there might be some selective basis for a particular segment of nucleic acid, or for an entire genome, arriving at a certain average base composition. In recent years evidence for the latter alternative has grown.[2,4,5,12,13,19-21] Changes in the base compositions of first and second codon positions provide an index of changes in protein-encoding potential (non-synonymous mutations). Changes in the base compositions of third codon positions provide an index of changes that do not change protein-encoding potential (synonymous mutations). Elementary information theory shows that information transmission potential is maximized when the four bases in a nucleic acid are present in equal proportions.[8,22] Thus, departures from 50% GC (downwards or upwards) are likely to progressively compromise the ability of genomes to transmit further information. Genomes with extreme variations from base equifrequency can be most helpful in displaying underlying genomic pressures. For example, at extreme genomic GC% values third codon positions serve mainly the information demands of a species, rather than of individual genes within members of that species.[21] As
GC% increases, AT% decreases, so GC% suffices to quantify both the
entity "GC-pressure" and, negatively, the entity
"AT-pressure." Similarly, AG% suffices to quantify both
"AG-pressure" and, negatively, "CT-pressure." GC%
increasing above 50% can be seen as a response to "upward
GC-pressure" rather than as a response to "downward
AT-pressure". GC% decreasing below 50% can be seen as a response to
"downward GC-pressure" rather than as a response to
"upward AT-pressure". In some circumstances it is convenient
to regard GC-pressure as increasing over the range 0% to 100%, so the
term must be understood in context.
|
|
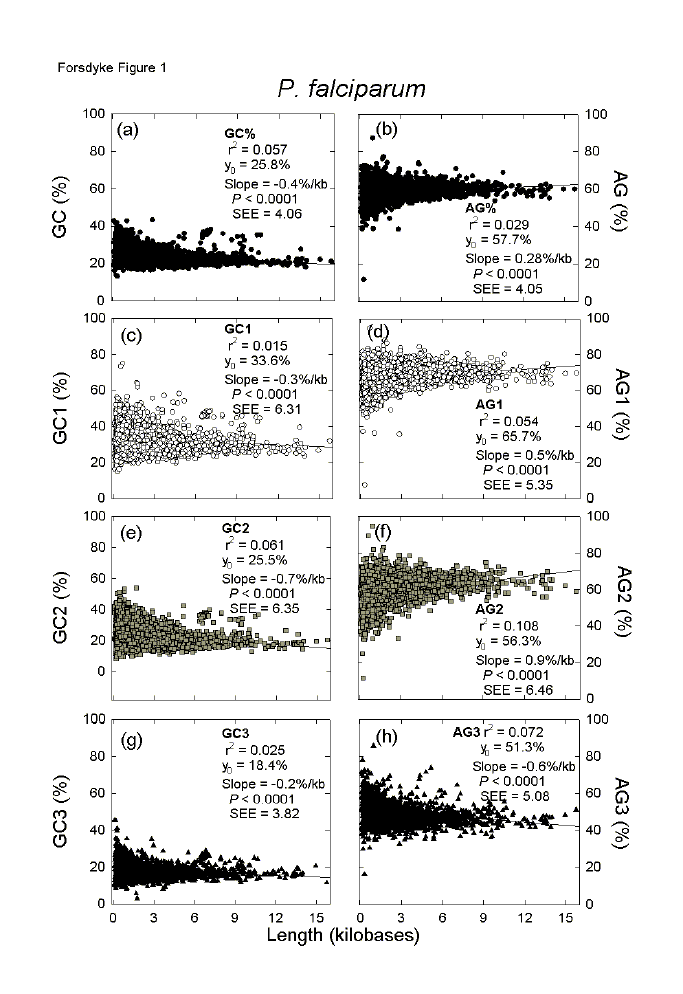
Most organisms have many small proteins and few very large ones. For an individual species, in plots of the base compositions of each protein-encoding region (ORF) against the corresponding lengths (kilobases), the multiple data points are distributed as rightward-pointing arrowheads.[12] Figure 1 shows this for 3772 genes of P. falciparum, each point corresponding to an individual gene. Many features of the distributions can be captured by first order linear regression analysis (Figures 1a, b). Although small proteins dominate the statistics, points at the tips of arrowheads that correspond to long proteins usually fit close to regression lines. second order regressions (not shown) offer little improvement. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Fig. 1. Variation of the base composition at different codon positions with ORF length in 3772 genes of the malaria parasite P. falciparum. Points were fitted to first order linear regression lines (r2 = adjusted square of the correlation coefficient; Y0 = intercept at the ordinate; P = probability that the slope is not significantly different from zero; seE = standard error of the estimate, which provides an index of the dispersion of points about the regression line and equals the standard error of the mean when slopes are zero). GC1, GC2, GC3, AG1, AG2 and AG3 refer to the base compositions (GC% or AG%) at different codon positions. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
As protein lengths increase there is a significant decline in GC% (a slope value of -0.4%/kb), which accounts for 6% of the variation between genes (r2 = 0.057), and a significant increase in AG% (a slope value of 0.28%/kb), which accounts for 3% of the variation (r2 = 0.029). These values indicate that the P. falciparum genome has been under downward GC-pressure (i.e. away from base equifrequency and towards low GC% values), and/or upward AG-pressure (i.e. away from base equifrequency and towards high AG% values). The "and/or" indicates that both pressures may be operating independently. However, there tends to be a reciprocal relationship between AG% and GC%, largely due to interchanges between A and C.[11,16] Thus, an increase in AG% ("purine-loading") can be at the expense of C, which is traded for A. So, if G% is constant, GC% will tend to decrease. Following, or as part of, an indel event, codons for putative placeholder amino acids would have appeared in a sequence.[23] A codon might have satisfactorily contributed to a base composition pressure immediately, or further mutation might have been required. Were base composition pressures accommodated without affecting the nature of encoded putative placeholder amino acids (changes primarily involving third codon positions), or were distinctive amino acids involved (changes primarily involving first and second codon positions)? For example, the amino acids whose single-letter designations spell "GARP" have codons whose first and second codon position maximally contribute to upward GC-pressure. Similarly the "GREK" amino acids can contribute to upwards AG-pressure. Whereas Figures 1a,b include all codon positions, Figures 1c-h relate to individual codon positions. Since the slope values (Figs. 1a,b) are largely contributed by first and second codon positions (Figs. 1c-h), the changes in base composition with length are likely to have involved distinctive amino acids, the codons of which were either directly inserted into sequences, or were derived by mutation from the originally inserted codons. In the case of GC%, all three positions contribute to the decline in GC% with ORF length, but the slope is greatest for second codon positions and 6% of the variation (r2 = 0.061) can be accounted for in terms of length differences (Fig. 1e). In the case of AG%, the contributions of first and second codon positions to the increase in AG% with ORF length are partly countermanded by the contributions of third codon positions (Fig. 1h). Nevertheless, the major contribution is from second codon positions where 11% of the variation (r2 = 10.8) can be accounted for in terms of length differences (Fig. 1f). Why were third codon position changes either meagre (GC%; Fig. 1g) or of a type that would countermand the non-synonymous mutation trend (AG%; Fig. 1h)? Other, even stronger, evolutionary pressures may have been operative. The inflexibility of third codon positions with respect to GC% differences is considered below. In the case of AG% differences, the negative slope in the case of third codon position bases suggests the operation of RNY-pressure - namely the translational pressure for third bases of codons to be pyrimidines (Y) rather than purines (R).[16] Delays in elongation or termination of proteins tend to stall ribosomes on mRNAs. Since stalling of protein synthesis on long polysomes (i.e. long mRNAs) would sequester more ribosomes than stalling on short polysomes (i.e. short mRNAs), thus potentially delaying overall protein synthesis, then genes corresponding to long ORFs might be particularly susceptible to RNY-pressure in organisms where the rate of protein synthesis was limiting growth rate.[24] Given the many data points corresponding to small ORFs and the generally modest slope values, intercepts at the ordinate (Y0 values) can provide an indication of overall base composition at each codon position. Thus, whereas the first and second codon positions contribute greatly to downward GC-pressure (Y0 = 33.6% and 25.5% respectively), the third codon positions make an even greater contribution (Y0 = 18.4%); all three negative slopes (Figs. 1c, e, g) show that this trend increases with gene length. Thus, it appears that, while small genes respond to downward GC-pressure, large genes have the capacity to respond more. Most organisms have average GC% values within the range 20% to 80%, so that the extremely low Y0 value and low slope value for third codon positions (Fig. 1g), suggests that the sequences are approaching functional AT-saturation and gene length cannot facilitate much of a further response to downward GC-pressure. Consistent with RNY-pressure being operative (first codon position is a purine, second codon position is any base, third codon position is a pyrimidine), first and second codon positions make the greatest contributions to upward AG-pressure (Y0 = 65.7% and 56.3% respectively; Figs. 1d, f); again, this is a trend that increases with gene length, so that large genes have the capacity to respond more to this pressure. The third codon positions of many genes are in slight purine excess (Y0 = 51.3%), but long genes tend to pyrimidine-load (negative slope; Fig. 1h) so that they would be in slight pyrimidine excess (in keeping with a special susceptibility of codons in long ORFs to RNY-pressure). |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
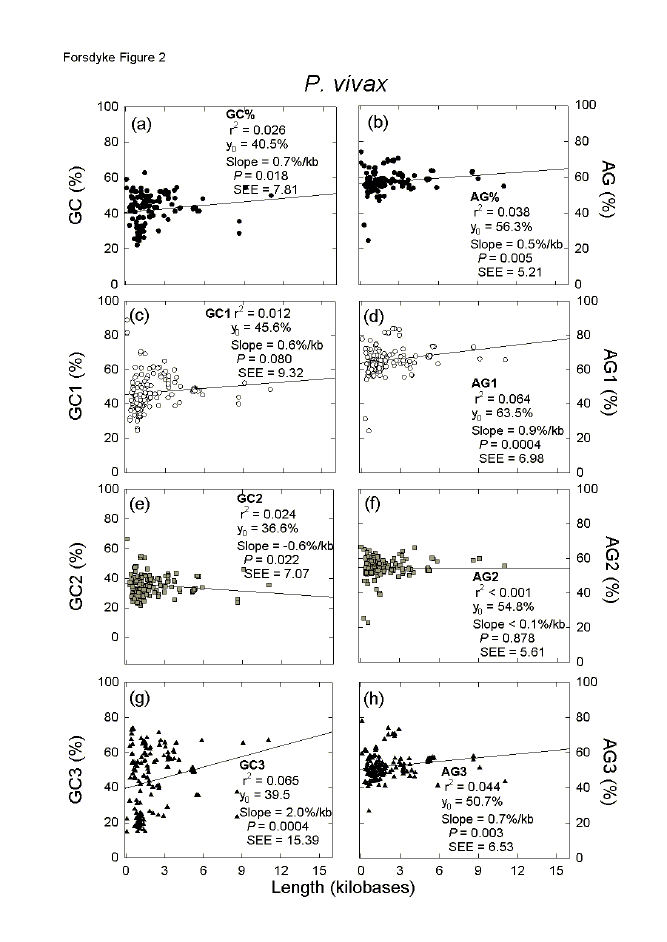
| Fig. 2. Variation of the base composition at different codon positions with ORF length in 179 genes of the malaria parasite P. vivax. For details please see the legend to Figure 1. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
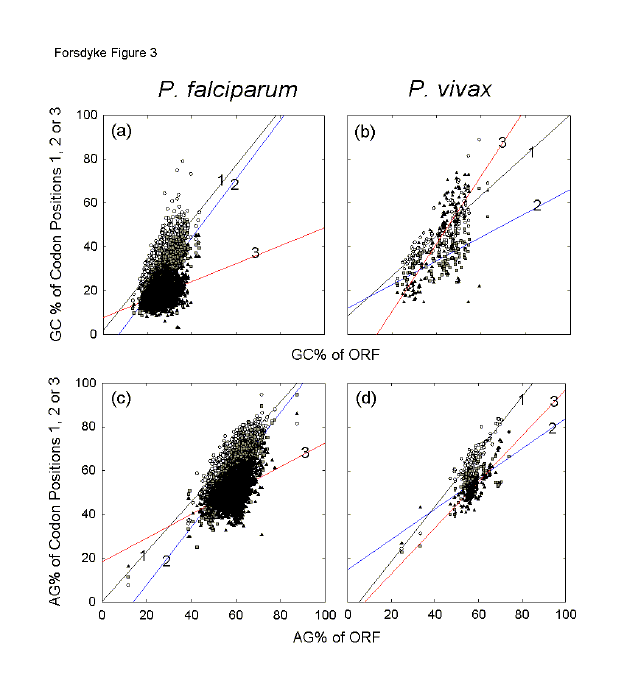
Another malaria parasite, P. vivax, appears to have been under less extreme GC-pressure, but under upward AG-pressure of the same order as P. falciparum (see Y0 values in Figs. 2a,b). Although relatively few genes were available for P. vivax in the CUTG, significant increases were observed in both GC% and AG% with increasing length of sequence (slope values of 0.7%/kilobase for GC% and 0.5%/kilobase for AG%; Figs. 2a,b). In contrast to P. falciparum, mainly the first and third codon positions are involved, and the primary amino acid-determining second codon positions play a minor role (Figs. 2c-h). For third codon positions there is a slight purine excess (Y0 = 50.7%), which long genes support (positive slope; Fig. 1h); thus in P. vivax there is not a general tendency to pyrimidine-load third codon positions. In this organism, which tends toward base compositional equifrequency, purine-loading trumps RNY-pressure. In P. falciparum the base of the arrowhead is very narrow in the case of the GC% of third codon positions (Fig. 1g), and this corresponds to a small standard error of the estimate (seE). Individual genes in P. falciparum vary little from each other in third codon position GC% values. In contrast, in P. vivax the base of the arrowhead for the GC% of third codon positions is very broad, corresponding to a much larger seE (Fig. 2g). Thus, individual genes in this organism can vary greatly from each other in third codon position GC% values, and it would be expected that an extreme response to base composition pressures, namely insertion of codons for placeholder amino acids, would be less necessary than in P. falciparum. The great differences between the two Plasmodium species in third codon position base compositions can be displayed by plotting the base compositions of individual codon positions against overall base compositions.[16,25] In general, the genes of P. falciparum have a very low GC% (Fig. 3a), and the genes of P. vivax have an intermediate GC% (Fig. 3b), but both species show the same order of purine-loading, and ORFs of AG% less than 50 (i.e. pyrimidine-loading) achieve this largely by virtue of third codon position values (see black triangles in Figs. 3c,d). As a function of genic GC%, third codon position GC% values of P. falciparum change little (low slope value; Fig. 3a), whereas third codon position GC% values of P. vivax change greatly (high slope value; Fig. 3b).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Fig. 3. Variation of the base compositions at different codon positions with the overall base compositions of the corresponding genes. The data are from P. falciparum as in Figure 1 (Figs. 3a, c), and from P. vivax as in Figure 2 (Figs. 3b, d). First order regression lines are shown with numbers indicating the corresponding codon positions. Symbols for different codon positions are as in Figures 1 and 2: open circles, first codon positions; grey filled squares, second codon positions; black-filled triangles, third codon positions. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
These data affirm that, in species under extreme GC-pressure (which may be upwards or downwards), third codon position GC% values tend to remain constant, serving the needs of the species rather than of individual genes, whereas in species with intermediate GC% values third codon position GC% values are more at liberty to vary, thus serving the needs of individual genes.[21] Accordingly, in P. falciparum the burden of responding to GC-pressure to serve the needs of individual genes rests primarily on first and second codon positions (Fig. 3a), so leading to changes in the nature of encoded amino acids. In P. vivax, third codon positions appear free to adopt the burden, and there is less pressure for amino acid change (low slopes for first and second codon positions; Fig. 3b). Values for AG% show similar, but not identical trends to those for GC% (Figs. 3c,d). In P. falciparum the slope value for third codon positions is low, perhaps reflecting a role of RNY pressure as suggested above. Thus, again, the burden of responding to AG-pressure rests primarily on first and second codon positions, so leading to changes in the nature of encoded amino acids. In P. vivax, the burden is largely carried by first and third codon positions. So there is less pressure for amino acid change. Thus P. vivax can respond to pressures for changes in base composition without necessarily changing the nature of its encoded amino acids, whereas for P. falciparum, this is mandatory. As will be shown below to usually apply to prokaryotes, there is a positive correlation of ORF length with GC% among genes of the eukaryote P. vivax (Fig. 2a), largely due to third codon position values (Fig. 2g). However, for many eukaryotic sequences the correlation is negative, [26-28] as is the case with P. falciparum (Fig. 1a).
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
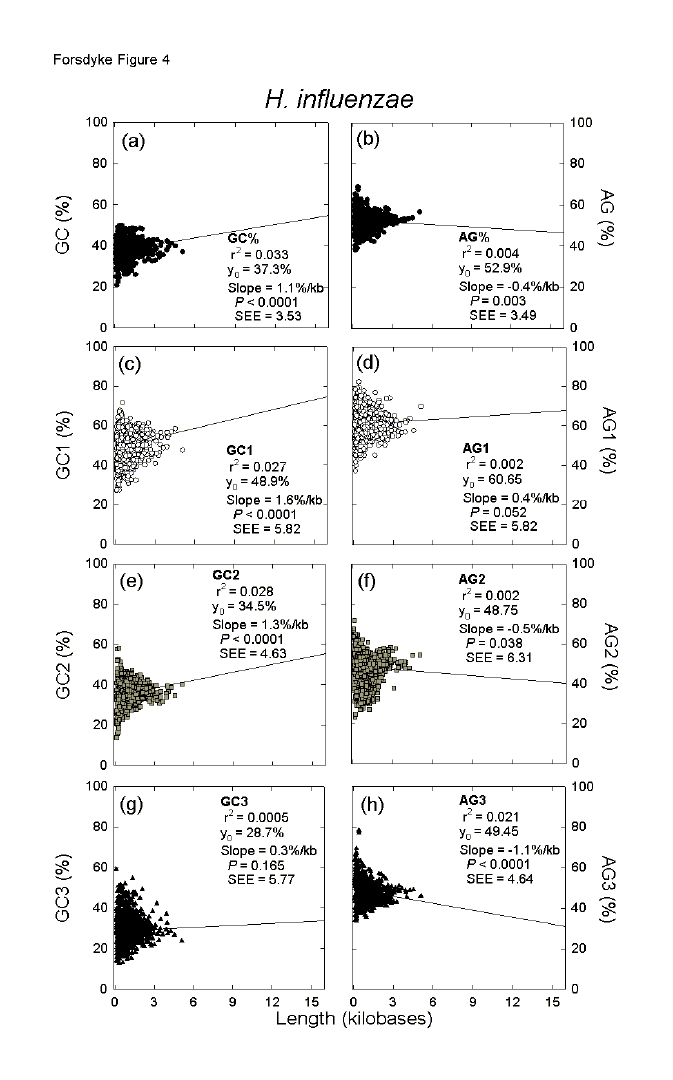
| Fig. 4. Variation of the base composition at different codon positions with ORF length in 1710 genes of the eubacterium Haemophilus influenzae. For details please see the legend to Figure 1. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
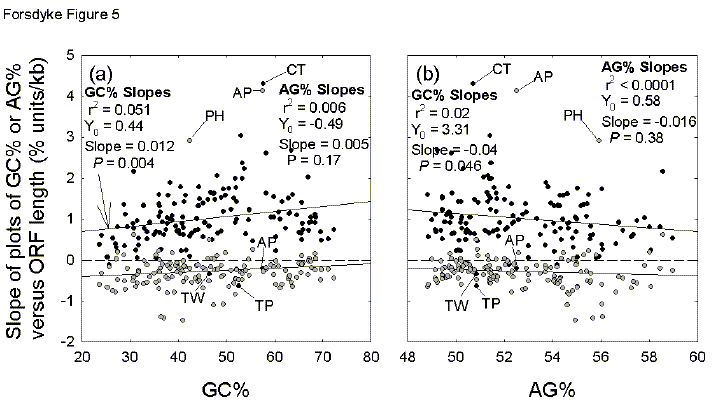
Fig. 5. Variation of base compositions with ORF lengths (slopes of plots as shown in Fig. 4a,b), as a function of genome base composition (a, GC%; b, AG%). Data are from 144 complete prokaryotic genomic sequences (125 eubacteria and 19 archaebacteria). Black-filled circles represent values of GC% / ORF length (slopes as in Fig. 4a). Grey-filled circles represent values of AG% / ORF length (slopes as in Fig. 4b). All species are included in the displayed statistics. Two letter abbreviations refer to points corresponding to species which deviate widely from the trends: AP, Aeropyrum pernix; CT, Chlorobium tepidum; PH, Pyrococcus horikoshii; TP, Treponema pallidum; TW, Tropheryma whipplei . |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
In Figures 4a and 4b there are two slope values (GC%/kb = 1.1 and AG%/kb =
-0.4), which summarize all codon positions of an individual prokaryote. In
Figure 5 the two corresponding slope values for the complete genomes of each of
144 prokaryotic species are summarized in terms of either the GC% (Fig. 5a) or
the AG% (Fig. 5b) of each species. Thus, whereas each point in Figure 4
corresponds to a gene, each point in Figure 5 corresponds to a species. With
five exceptions (Aeropyrum pernix, Legionella pneumophila, Treponema
pallidum, Tropheryma whipplei, Ureaplasma parvum), GC% slope
values (%/kb) are positive with an average of 1.03se0.06 %/kb (P
<0.0001 that not significantly different from zero). With two major
exceptions (Aeropyrum pernix, Pyrococcus horikoshii), AG% slope
values are close to zero or negative with an average of -0.26se0.05 %/kb (P
< 0.0001). Thus, relative to short ORFs, long ORFs tend to contribute
positively to GC% and negatively to AG%. Do the extents of these contributions
vary with species base composition?
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
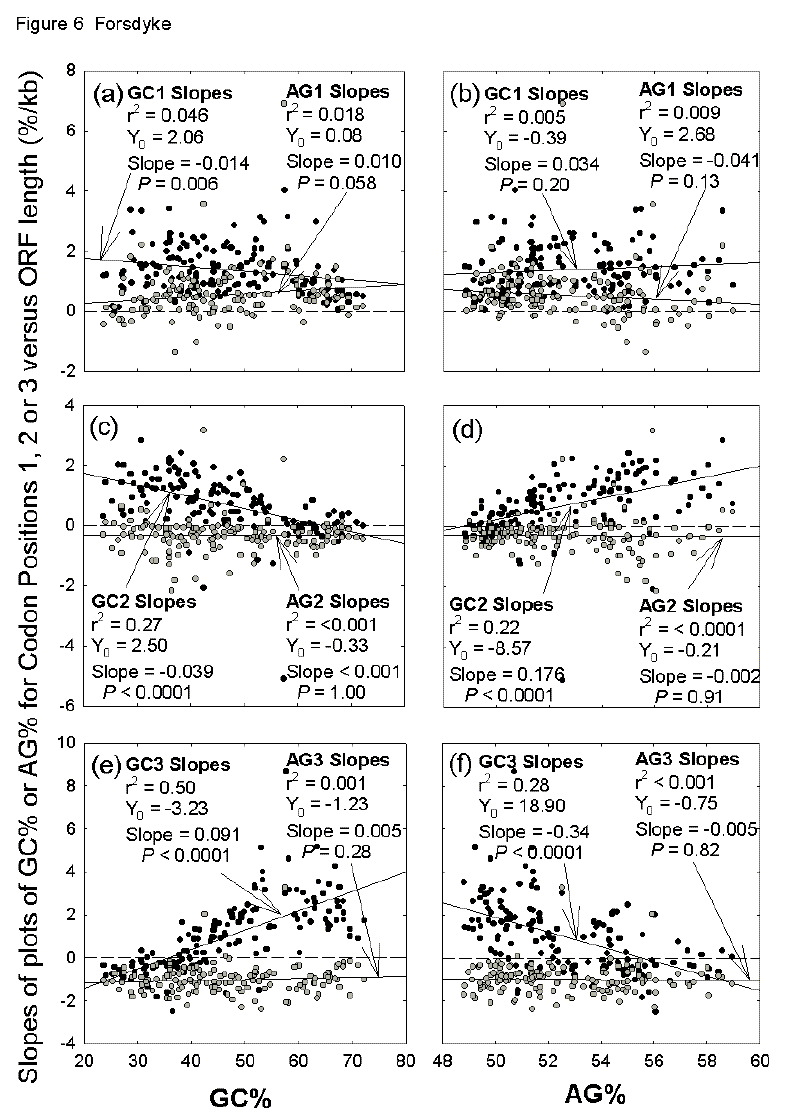
Fig. 6. Variation of the base composition at different codon positions with ORF lengths (slopes of plots as shown in Figures 4c-h), as a function of genome base composition (a, c, e, GC%; b, d, f, AG%). Other details are as in Figure 5. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
In Figure 4 there are six slope values for different codon positions (Figs. 4c-h). In similar fashion to Figure 5, these are displayed for the 144 prokaryotic species in Figure 6. Taking all codon positions into account, slope values for AG% (AG%/kb) are generally negative (Fig. 5). However, indicating the importance of RNY-pressure, values at first codon positions are generally positive (Figs. 6a, b; average = 0.55se0.07; P < 0.0001), values at second codon positions are generally slightly negative (Figs. 6c, d; average = -0.33se0.05; P <0.0001), and values at third codon positions are generally strongly negative (Figs. 6e, f; average = -1.01se0.06; P <0.0001). Again, whatever the codon position, these values do not vary significantly with species base compositions. Although, taking all codon positions into account, slope values for GC% (GC%/kb) are positive (Fig. 5), in the case of second codon positions, values become zero or negative in organisms with a high overall GC% (Fig. 6c) and a low overall AG% (Fig. 6d) The converse applies, and more dramatically so, in the case of third codon positions (Figs. 6e, f). Thus, the increasing role of gene length in contributing positively to the overall GC% of a prokaryote as species GC% increases (Fig. 5a), is dealt with in different ways be different prokaryotic species. In low and medium GC% species, increases in open reading frame GC% as the corresponding encoded proteins increase in length are largely accounted for by the base compositions at first and second codon positions. In high GC% species first, and especially third, codon positions play this role. So in low and medium GC% prokaryotes the increase in GC% in long open reading frames is contributed by distinct placeholder amino acids with GC-rich first and second codon positions (amino acid-determining). In high GC% prokaryotes the further increase in GC% in long open reading frames allows flexibility in the nature of placeholders, which mainly contribute by virtue of the GC-richness of their first and third codon positions (non-amino acid-determining). In Figure 6c the downward slope crosses the abscissa at a GC% value of 64, whereas in Figure 6e the upward slope crosses the abscissa at a GC% value of 35. These values approximate to those previously identified (68 and 38, respectively) as the GC% values at which there are transitions from primarily serving genic demands to primarily serving species demands.[21] Incomplete Genomes Confirm Complete F or reasons given above (see Data), a further 377 sequences from incompletely sequenced prokaryotic genomes were added to the above sequences from 144 completely sequenced genomes. The various linear regression parameters, as in Figures 5 and 6, are shown in Table 1. While there are some differences from the results obtained when only completely sequenced genomes were studied, the essential observations are confirmed, suggesting that they apply to prokaryotes in general. However, in the case of GC% slopes the slope values for first and second codon positions tend to cancel out the slope values for third codon positions, so that slope values for all codon positions are not significantly different from zero (c.f. Fig. 5a). In these studies no major differences were detected between archaebacteria and eubacteria.
Protein Length Constrained in Thermophiles AG% (purine-loading) increases with optimum growth temperature, whereas GC% tends to decrease.[10-13,17] Since AG-pressure can increase protein lengths by virtue of interdomain insertions of purine-rich simple sequence elements,[3,9] which should be extruded as loops from protein structures, it would be predicted that homologous proteins would be longer in thermophiles than in mesophiles. However, it is probable that thermophilic proteins achieve a greater compactness than mesophilic proteins by decreasing the length and number of external loops.[29] Consistent with this, ORFs tend to be shorter in thermophiles than in mesophiles.[30] Of the above 144 prokaryotic species for which complete genomic sequences are available, the optimum growth temperatures of 108 were available in the PGTdb. Of the above 521 prokaryotic species for which either complete or incomplete sequences (containing 20 or more ORFS) were available, the optimum growth temperatures of 293 were available in the PGTdb. The slope values (%/kb) as recorded above, were plotted as a function of optimum growth temperatures, which ranged from 0oC to 101.5oC. Because there was an overrepresentation of species with optimum growth temperatures of 37oC or less, slope values were also plotted for a narrower temperature range (38oC to 101.5oC). Some statistical parameters from the first order linear regression plots (not shown) are given in Table 2.
Whatever the codon position, whatever the temperature range, and whether or not incomplete sequences are included, slopes of plots of AG% against ORF lengths (kb) do not change significantly as optimum growth temperatures increases. This implies that prokaryote thermophiles, while they may purine-load, do not concomitantly increase protein length to accommodate this. Thermophiles purine-load within the constraints of ORF length and would not seem, in general, to employ interdomain regions for this purpose. For complete genomes, over a broad temperature range slopes of plots of GC% for all codon positions against ORF lengths decreased slightly as optimum growth temperatures increased (r2 = 0.034; P = 0.031), but this was not significant when incomplete genomes were included (r2 = 0.007; P = 0.09), nor when broken down to individual codon positions. However, over a narrower temperature range (38oC to 101.5oC) the decrease was highly significant, both with complete genomes alone (r2 = 0.436; P = 0.0008), and when incomplete genomes were added (r2 = 0.135; P = 0.002). All three codon positions contribute to the decrease, with third codon positions usually making the greatest and most significant contribution (r2 = 0.236; P = 0.01 for complete genomes; r2 = 0.09; P = 0.01 when incomplete sequences were included). The regression lines sloped downwards to cross the abscissa at a point corresponding to a growth temperature of approximately 100oC (data not shown). Thus, species with low growth temperatures can respond to GC-pressure by increasing ORF lengths, possibly employing interdomain regions for "GC-loading" (e.g. Figs. 4a,c,e), but this is countermanded in species with high growth temperatures. In other words, thermophiles tend not to load GC, perhaps again reflecting constraints on loop formation and ORF lengths at high temperatures, but also perhaps reflecting the reciprocal relationship between AG-pressure (high in thermophiles) and GC-pressure. Base Composition and Chain-Terminating Codons S ince chain-terminating codons are GC-poor, in a random GC-rich sequence there should be a deficit of chain-terminating codons (UAA, UAG, or UGA) and hence longer potential ORFs.[31] Indeed, the positive correlation between GC% and ORF length noted for many prokaryotes (Fig. 4a)[27,32] would seem consistent with this. However, although the extent of the positive correlation increases with species GC%, the correlation is general, occurring both in low GC% and high GC% species (Fig. 5a). Thus, deficit of a small subset of GC-poor chain-terminating codons is unlikely to be a major factor in the correlation.Long genes, in general, may have such different functions from short genes, in general, that different amino acids are required. For example, in eukaryotes housekeeping genes tend to be shorter than tissue-specific genes; this is attributed to the "functional architectures" of their proteins, which require fewer functional domains.[33] In prokaryotes, membrane proteins rich in hydrophobic amino acids tend to be shorter.[32] So, by default, long prokaryotic genes might happen to need, for the optimal functioning of their encoded proteins, more GC-rich codons. Indeed, this would appear consistent with long genes from low or intermediate GC% prokaryotes contributing to increased GC-pressure by virtue of second (amino acid determining) codon positions. However, it does not explain why long genes from GC-rich prokaryotes do not also contribute to increased GC-pressure by virtue of second codon positions (Fig. 6c). On balance, the data support the placeholder hypothesis, rather than the function hypothesis. But why would distinct amino acids be involved in the case of low and intermediate GC% prokaryotes? Placeholder amino acids with GC-rich first and second codon positions ("GARP") might interfere less with the functions of their proteins than others. For example beta turns, usually occurring at protein surfaces, often contain proline (P) and glycine (G), which may be accompanied by, or form part of, simple sequence repeats.[23] If this argument applies to prokaryotic proteins it might also apply to eukaryotic proteins, and there should again be a positive relationship between ORF length and GC%. However, the relationship is usually negative (Fig. 1).[26-28] Here purine-loading may be of more importance. In the extreme case of P. falciparum, increased purine-loading with ORF length accounts for 11% of the variation in second codon position purine levels(Fig. 1f) and purine-loading is seen in many simple sequence elements.[1-3] Thus, purine-loading appears to dominate and, in view of the reciprocal relationship between AG% and GC%, the negative relationship between ORF length and GC% may be secondary to this. Eukaryotic proteins are generally longer than prokaryotic proteins,[34] their ORFs are preferentially loaded with long purine tracts,[35] and their simple sequence elements can confer intragenic codon bias.[9,36] Again, in view of the reciprocal relationship (increased AG% implies decreased GC%), purine-loading might be driving the generally negative relationship between ORF length and GC% in eukaryotes. Most results of this study are explained in terms of two major factors, base
composition pressures and RNY-pressure. While the causes of base composition
pressures remain contentious, the pressures themselves can be dealt with as
abstract entities. Insertion of placeholder amino acids would be an extreme
response to base composition pressures. The best evidence supporting this has
derived from organisms that deviate strongly from base equifrequency. In this
study less strongly deviant species have also been considered. Despite the
distinct possibility that the putative placeholders do not contribute positively
to the specific functions of the proteins containing them, in prokaryotes of low
and intermediate GC% placeholder-choice is not random. Placeholder amino acids
with GC-rich first and second codon positions might interfere less with the
functions of their proteins than others. For example beta turns, usually occurring
at protein surfaces, often contain proline and glycine, which may be accompanied
by, or form part of, simple sequence repeats.[23] However, certain
prokaryotes (e.g. Aeropyrum pernix) deviate dramatically from the general
trend (Fig. 5).[12] The possibility of annotation errors has been
referred to.[15] Nevertheless, following Bateson's admonition to
"treasure your exceptions,"[37] it is possible that focused
studies of these organisms will be highly informative. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
![]()
References
1. Pizzi E, Frontali C. Low-complexity regions in Plasmodium falciparum proteins. Genome Res 2001; 11:218-229
2. Forsdyke DR. selective pressures that decrease synonymous mutations in Plasmodium falciparum. Trends Parasitol 2002; 18:411-418
3. Xue HY, Forsdyke DR. Low complexity segments in Plasmodium falciparum are primarily nucleic acid level adaptations. Mol Biochem Parasitol 2003; 128:21-32
4. Forsdyke DR, Mortimer JR. Chargaff's legacy. Gene 2000; 261:127-137
5. Forsdyke DR. Functional constraint and molecular evolution. Nature Encyclopedia of Life Sciences 2002; 7:396-403
6. Schaap T. Dual information in DNA and the evolution of the genetic code. J Theor Biol 1971; 32:293-298
7. Ball LA. Implications of secondary structure in messenger RNA. J Theor
Biol 1972; 36:313-3208. Wan H, Wootton JC. A global compositional complexity measure for biological sequences: AT-rich and GC-rich genomes encode less complex proteins. Comput Chem 2000; 24:71-94
9. Cristillo AD, Mortimer JR, Barrette IH, Lillicrap TP, Forsdyke DR. Double stranded RNA as a not-self alarm signal: to evade, most viruses purine-load their RNAs, but some (HTLV-1, EBV) pyrimidine-load. J Theor Biol 2001; 208:475-491
10. Lao PJ, Forsdyke DR. Thermophilic bacteria strictly obey Szybalski's transcription direction rule and politely purine-load RNAs with both adenine and guanine. Genome Res 2000; 10:228-236
11. Lambros RJ, Mortimer JR, Forsdyke DR. Optimum growth temperature and the base composition of open reading frames in prokaryotes. Extremophiles 2003; 7:443-450
12. Paz A, Mester D, Baca I, Nevo E, Korol A. Adaptive role of increased frequency of polypurine tracts in mRNA sequences of thermophilic prokaryotes. Proc Natl Acad Sci USA 2004; 101:2951-2956
13. Friedman R, Drake JW, Hughes AL. Genome-wide patterns of nucleotide substitution reveal stringent functional constraints on the protein sequences of thermophiles. Genetics 2004; 167:1507-1512
14. Nakamura Y, Gojobori T, Ikemura T. Codon usage tabulated from the international DNA sequence databases: status for the year 2000. Nucleic Acids Res 2000; 28:292
15. Skovgaard M, Jensen LJ, Brunak S, Ussery D, Krogh A. On the total number of genes and their length distribution in complete microbial genomes. Trends Genet 2001; 17:425-428
16. Mortimer JR, Forsdyke DR. Comparison of responses by bacteriophage and bacteria to pressures on the base composition of open reading frames. Applied Bioinformatics 2003; 2:47-62
17. Lobry JR, Chessel D. Internal correspondence analysis of codon and amino acid usage in thermophilic bacteria. J Appl Genet 2003; 44:235-261
18. Osawa S, Jukes TH, Muto A, Yamao F, Ohama T, Andachi Y. Role of directional mutation pressure on the evolution of the eubacterial genetic code. Cold Spring Harb Symp Quant Biol 1987; 52:777-789
19. Forsdyke DR. The Origin of Species, Revisited. Montreal: McGill-Queen's University Pr. 2001
20. Chen SL, Lee W, Hottes AK, Shapiro L, McAdams HH. Codon usage between genomes is constrained by genome-wide mutational processes. Proc Natl Acad Sci USA 2004; 101:3480-3485
21. Lee S-J, Mortimer JR, Forsdyke DR. Genomic conflict settled in favour of the species rather than of the gene at extreme GC% values. Applied Bioinformatics 2004; 3:219-228
22. Khinchin AI. Mathematical Foundations of Information Theory. New York: Dover Publications 1957
23. Chang MSS, Benner SA. Empirical analysis of protein insertions and deletions determining parameters for the correct placement of gaps in protein sequence alignments. J Mol Biol 2004; 341:617-631
24. Kurland CG. Major codon preference: theme and variation. Biochem Soc Trans 1993; 21:841-846
25. Muto A, Osawa S. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci USA 1987; 84:166-169
26. Marais G, Duret L. Synonymous codon usage, accuracy of translation, and gene length in Caenorhabditis elegans. J Mol Evol 2001; 52:275-280
27. Xia X, Xie Z, Li W-H. Effects of GC content and mutational pressure on the lengths of exons and coding sequences. J Mol Evol 2003; 56:362-370
28. Wang H-C, Singer GAC, Hickey DA. Mutational bias affects protein evolution in flowering plants. Mol Biol Evol 2004; 21:90-96
29. Russell RJM, Ferguson JMC, Hough DW, Danson MJ, Taylor GL. The crystal structure of citrate synthase from the hyperthermophilic archaeon Pyrococcus furiosus at 1.9 Å resolution. Biochemistry 1997; 36:9983-9994
30. Tekaia F, Yeramian E, Dujon B. Amino acid composition of genomes, lifestyles of organisms, and evolutionary trends: a global picture with correspondence analysis. Gene 2002; 297:51-60
31. Forsdyke DR. sense in antisense? J Mol Evol 1995; 41:582-586
32. Hooper SD, Berg OG. Gradients in nucleotide and codon usage along Escherichia coli genes. Nucleic Acids Res 2000; 28:3517-3523
33. Vinogradov AE. Compactness of human housekeeping genes: selection for economy or genomic design? Trends Genet 2004; 20:248-253
34. Brocchieri L. Environmental signatures on proteome properties. Proc Natl Acad Sci USA 2004; 101:8257-8258
35. Yagil G. The over-representation of binary DNA tracts in seven sequenced chromosomes. BMC Genomics 2004; 5:19 http://www.biomedcentral.com/1471-2164/5/19
36. Desai D, Zhang K, Barik S, Srivastava A, Bolander ME, Sarkar G. Intragenic codon bias in a set of mouse and human genes. J Theor Biol 2004; 230:215-225
37. Punnett RC. William Bateson. The Edinburgh Review 1926; 244:71-86
This paper was submitted to the journal Genetics (4 sept 2004), but the two anonymous reviewers advised the handling editor, Shozo Yokoyama, to reject. Applied Bioinformatics is unfortunately not widely read. While unaware of some of the points made here, a new paper (Sabath et al. 2013) is generally supportive of our case that genomic streamlining is adaptive.
Sabath E, Ferrada E, Barve A, Wagner A (2013) Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biology and Evolution 5, 966-977.
Return to Malaria Page Click Here
Go to Bioinformatics Index Click Here
Go to Home Page Click Here
![]()
Placed on the Internet in July 2005 and last edited 08 Nov 2020 by Donald Forsdyke