D. R. FORSDYKEJournal of Biological Systems (2004) 12, no. 3, 261-271
|
REGIONS OF
RELATIVE GC% UNIFORMITY ARE RECOMBINATIONAL ISOLATORS
D. R. FORSDYKEJournal of Biological Systems (2004) 12, no. 3, 261-271
|
![]()
Among species within a phylogenetic group, genomic GC% values can cover a wide range that is particularly evident at third codon positions. However, among genes within a genome, genic GC% values can also cover a wide range that is, again, particularly evident at third codon positions. Individual genes and genomes each have a "homostabilizing propensity" to adopt a relatively uniform GC%. Each gene (a "microisochore") occupies a discrete GC% niche of relatively uniform base composition amongst its fellow genes, which can collectively span a wide GC% range. Homostabilization serves to recombinationally isolate both genome sectors (facilitating gene duplication and differentiation) and genomes (facilitating genome duplication and differentiation; e.g. speciation). Although they may sometimes be in conflict, the individualities of genomes, and of genes within those genomes, are separately sustained by a common mechanism, uniformity of GC%. The protection against inadvertent recombination afforded by GC% differentiation is, in the general case, a prerequisite for phenotypic differentiation. |
Key
words:
Base composition; Codon position; Gene duplication; Gene homostabilizing
propensity; Selfish genes; Speciation
![]()
|
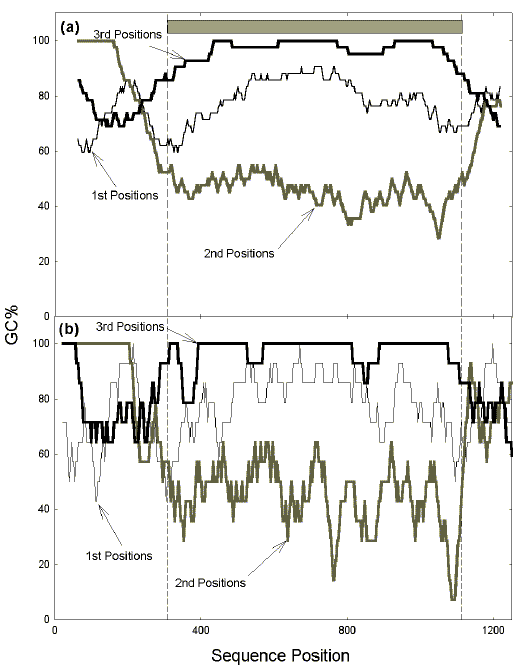
Prior to the emergence of modern sequencing technologies in the 1970s, various physical techniques demonstrated small segments of distinct average GC% in the genomes of prokaryotes and their viruses. Following extensive hydrodynamic shearing to produce subgenome-sized duplex fragments, the 48 kilobase lambdaphage genome was resolved into six segments of differing densities on salt gradients. The segments were "homogeneous internally and the boundaries between segments rather sharp." The densities could be related to the average GC% values of the segments since the greater these values, the greater the densities [24]. A more sensitive technique, thermal denaturation spectrophotometry, revealed thirty four "gene sized" segments in unsheared lambdaphage DNA, each of relatively uniform GC% [26]. Similar thermal denaturation studies, and later direct sequence analyses, led to the observation by Wada et al. [27, 30] that each gene within a prokaryotic genome has a "homostabilizing propensity," so that "every base in a codon seems to work cooperatively towards realizing the gene's characteristic value of (G + C) content". This was elegantly shown by Bibb et al. in 1984 [3]. They plotted the average GC% values of every third base in 120 base windows in the sequences of various bacteria. Three plots were generated, the first beginning with the first base of the sequence (i.e. bases in frame 1, 4, 7, etc.), the second beginning with the second base of the sequence (i.e. bases in frame 2, 5, 8, etc.), and the third beginning with the third base of the sequence (i.e. bases in frame 3, 6, 9, etc.). In certain regions GC% values were relatively constant for all frames (Fig. 1a). These regions of constant GC% corresponded to genes. Figure 1b shows that the constancy is evident with windows equivalent to only 14 codons. Constancy is most in the case of the codon position of least importance to amino acid determination (third position). Constancy is least in the case of the codon position of most importance to amino acid determination (second position). The greater GC% fluctuations in the case of first and second codon positions are likely to reflect local needs for the encoding of distinct amino acids.
The "homostabilizing propensity" allows a prokaryotic gene to maintain a distinct GC%, relatively uniform along its length, which can differentiate it from other genes in the same genome. Various studies [17, 23] indicate some colocalization of genes of a common GC% in prokaryotes. The relationship of these regions of relatively uniform GC% to the large regions in eukaryotic genomes that Bernardi et al. [2, 6] named "isochores" has been unclear. The purpose of the present communication is to draw attention to the growing evidence for functional similarities between these regions. |
|
The density gradient method [24] was also applied to eukaryote genomes. Thus, isochores were described as large DNA segments that could be identified on the basis of their distinct densities [2, 6]. Prior to resolution on salt gradients, eukaryotic DNA was sheered to produce duplex segments of about 300 kilobases. This method of assessing GC% distinguished one large segment from another, and largeness became a defining property of isochores. Accordingly, they were not observed in prokaryotes. Yet the gene-sized regions with a "homostabilizing propensity" that had been observed in prokaryotes, were also observed in eukaryotes [25, 28]. Thus, every gene, be it prokaryotic or eukaryotic, has a propensity to stabilize its GC% value. Accordingly, classical isochores can be viewed as collections of homostabilizing regions within a region of common GC%. Classical isochores would not be detected in prokaryotic genomes because the homostabilizing regions are not generally collected into large regions of similar GC% [17, 23]. It is suggested here that, whatever their size, differentiation of regions by virtue of the uniformity of their GC% plays one major role - recombinational isolation. In the light of this common function, it is suggested that classical isochores should henceforth be considered as "macroisochores." The smaller regions with the homostabilizing propensity should be considered as "microisochores." Together, they constitute a general isochore group defined solely in terms of regional base composition uniformity. |
3. "Universal" codon position GC% plots
|
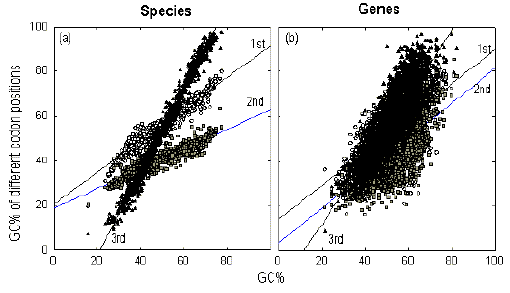
As shown in Figure 1, although each codon position is relatively uniform in its average GC% value within a gene, different codon positions within a gene have different average GC% values. It has been observed that this codon position differentiation is evident both when genes within a genome are compared, and when genomes within a phylogenetic group are compared [5, 29].
Figure 2a shows a plot of average GC% values of different codon positions of the set of available gene sequences from each of 898 prokaryotic species against the average genomic GC% of that species [21]. As has been shown for a smaller number of species [22], the slope for the second codon position, which is most constrained by the need to specify amino acids, is lowest. The slope for the third codon position, which is least constrained by the need to specify amino acids, is greatest. Similar plots are obtained with various eukaryotic groups. Thus, for the set of sequenced genes of a species, the third codon position appears to amplify either a low genomic GC% by exceeding the first and second codon positions in its AT-richness, or a high genomic GC% by exceeding the first and second codon positions in its GC-richness. This result might be viewed as consistent with each genome being a collection of genes within a region of relatively uniform GC%. However, almost identical patterns of regression lines are obtained when the GC% values of individual genes within a genome, be it prokaryotic or eukaryotic, are plotted. Thus, whereas in Figure 2a each point represents a species within a phylogenetic group, the same result is obtained when each point represents a gene within a species (Figure 2b). This led Wada et al. in 1991 [29], and others [5], to describe the plots as "universal" suggesting that the constraints operating on the three codon positions "must have general biological meanings in relation to the DNA/RNA and protein functions." |
4. Function of homostability regions
|
Since prokaryotes and eukaryotes are considered to have evolved from a common ancestor, the question arises as to whether that ancestor had isochores. As long as isochores were considered a distinctive feature of eukaryotes, it was appropriate to ask whether the ancestor had isochores that were subsequently lost by prokaryotes during or after their divergence from the eukaryote lineage (isochores-early)? Alternatively, did the ancestor not have isochores, which were therefore freshly acquired by the eukaryotic lineage after its divergence from the prokaryotic lineage (isochores-late)? If, as argued here, modern prokaryotes and eukaryotes both have isochores, then this makes it likely that the ancestor also had isochores, although we do not know whether they were small (microisochores) or large (macroisochores). Nevertheless, their conservation over this long evolutionary period supports Wada's contention that regions of uniform GC% have a fundamental biological meaning. What could that meaning be? At the outset, the possibility of a role in recombination was entertained. In 1968 Skalka et al. [24] suggested that if base "composition and function are indeed related," then segments of relative GC% uniformity [i.e. segments demarkated by differences in GC%. DRF 2009] would appear "not to encourage recombination" between functional units. However, the possibility of highly localized mutational biases that would favour a regional GC% was also entertained. Thus, Skalka et al. [24] acknowledged that each segment might have "its own set of critical nucleotide sequences, each set adapted to a different mutational habit" that would determine the segment's GC%. No evidence for such a highly localized "mutational habit" has since emerged. In 1976 Wada et al. [30] found it "hard, if not impossible, to believe" that the homostabilizing regions reflected a fundamental characteristic of the genetic code itself. Rather, the regions must play "an important part somewhere in the biological process within which the DNA is closely related-- From the size of the homostability region, recombination might be one possible process --". |
5. Function of classical isochores
|
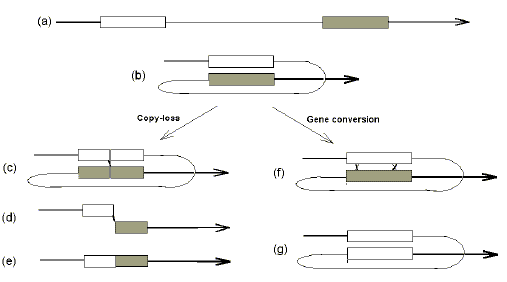
Further evidence for a relationship between GC% and recombination arose from studies of duplicate genes in eukaryotes. Based on sequence similarities, genes are sometimes considered to have arisen by the intragenomic duplication of a single ancestral gene. The duplicates may then undergo either concerted or divergent evolution. When the duplicates differ little in sequence (and hence, function; e.g. rRNA genes) then recombination-mediated gene conversion is held to assist the maintenance of sequence uniformity (concerted evolution). The survival of the duplicate is maintained by natural selection if such duplication is advantageous (e.g. the organism can more readily respond to demands for large quantities of gene product). However, the survival of the duplicate is always at risk since intragenic recombination between paralogues might eliminated one copy (copy-loss; Fig. 3). When functional differentiation of a duplicate is necessary for it to be selected (divergent evolution), there is the danger that, before natural selection can operate, recombination-mediated gene conversion will reverse any incipient differentiation, or intragenic recombination between paralogues will result in copy-loss.
In the case of duplicate eukaryotic genes that have diverged in sequence, Matsuo et al. [18] noted that divergence was greatest at third codon positions, usually involving a change in GC% [32]. Thus, there was a codon bias in favour of the positions of least importance for the functional differentiation that would be necessary for the operation of natural selection. Where amino acids had not changed, different gene copies used different synonymous codons. It was proposed that the GC% change was an important "line of defence" against homologous recombination between the duplicates. Thus, recombinational isolation of the duplicate (largely involving third codon position GC% differences) would protect the duplicate so allowing time for (i) functional differentiation (largely involving first and second codon position differences), and hence (ii) preservation by natural selection [20]. In the general case, isolation would precede functional differentiation, not the converse. GC% differentiation, largely involving third codon positions, would precede functional differentiation, largely involving first and second codon positions. The third position GC% differentiations would then be preserved by natural selection acting through the first and second positions. Many first and second positions would not change since it is likely that the diverged genes would retain some similar functions. Thus, it is likely that third position differentiations would play a critical role in initiating the isolation process. Matsuo et al. [18] also proposed that "one possible way to maintain a codon bias would be to deposit members of a gene family in different so-called isochores, which are defined as long (>300 kb) genomic DNA segments of a characteristic G + C content". Thus, a classical isochore might have arisen as a random fluctuation in base composition in a genomic region such that a product of a gene duplication, which had been deposited in that region, was able to survive for a sufficient number of generations to allow it to differentiate both recombinationally (i.e. change third codon positions) and functionally (i.e. change and first and second codon positions). The linked regional base compositional fluctuation would then have "hitch-hiked" through the generations on the duplicate (if the latter were later favoured by natural selection) [13]. Once a classical isochore was established then, by virtue of its GC%, it would provide a safe-haven for future incipient gene duplicates. At the moment of initial deposition, the probability of recombination between identical duplicates would be decreased by the GC% differences between flanking regions of the source isochore and the new host isochore (Fig. 3). Later changes in GC% within the deposited gene would enhance its recombinational isolation if they happened to make it further match its host isochore. On this basis it would be predicted that, if a gene from one classical isochore were copied or transposed to another classical isochore of different GC%, then the gene would preferentially accept mutations converting its GC% to that of the new host isochore (i.e. over time, organisms with favourable GC% mutations would be likely to leave more fertile offspring than organisms without such mutations). Indeed, there is recent evidence supporting this. The sex chromosomes (X and Y) tend not to recombine except in a small region known as the "pseudoautosomal" region. Transfer of a gene from a non-recombining part of a sex chromosome to the pseudoautosomal region forces the gene rapidly to change its GC% value. This has led to a growing appreciation that "recombination explains isochores" [19]. Furthermore, Iwase et al. [15] have suggested that "recombination suppression is somehow related to long range mosaic structures in the genome in terms of the GC content". The proposed antirecombination role of differences in GC% would require that, unless representing concertedly evolving multicopy genes, microisochores sharing a common macroisochore (i.e. they have a similar GC%) have other sequence differences that are sufficient to prevent recombination between themselves. |
6. Why are codon position GC% plots "universal"?
|
The power to recombine is fundamental to all life forms because, for a variety of reasons, it is advantageous [11]. However, the same power threatens to homogenize the genes within a genome, and to homogenize the genomes of allied species within a phylogenetic group, so countermanding evolution both within a species and between species. Thus, functional differentiation, be it between genes in a genome, or between genomes in a phylogenetic group, must, in the general case, be accompanied or preceded by the establishment of recombinational barriers. If differences in GC% can serve to recombinationally isolate genome sectors, so facilitating gene duplication, then differences in GC% might serve to recombinationally isolate genomes, so facilitating genome duplication (e.g. speciation). The very same forces that drive diversification among a set of genes within a genome, might also drive diversification among a set of genomes within a phylogenetic group. This predicts that codon position GC% plots will be "universal." Just as, within a genome, genic GC% values cover a wide range that is most evident at third codon positions, so, within a phylogenetic group, genomic GC% values should cover a wide range that is most evident at third codon positions (Fig. 2). Thus, whatever was causing a dispersion of GC% values among genes (e.g. the need to avoid intergenic recombination), might also be causing a dispersion of GC% values among genomes (e.g. the need to avoid intergenomic recombination). There might then be a conflict between the GC% needs of individual "selfish" genes, and the GC% needs of the "selfish" genomes within which these genes reside. In this context, as pointed out by Williams in 1966, "gene" means any portion of chromosomal material that has the potential to last for enough generations to serve as a unit of natural selection; this requires that it not be easily disruptable by recombination [4]. It is shown elsewhere that in species with extreme genomic GC% values there is indeed a conflict, which is settled in favour of the species [31]. It is also shown elsewhere that differences in GC% would, in the general case, provide a degree of reproductive isolation sufficient to facilitate the initiation of species divergence into two lines [7-12]. Usually, this initial, gene-independent, interspecies barrier (hybrid sterility) would be replaced later by gene function-dependent barriers (hybrid inviability, prezygotic isolation), so that there would be less pressure to retain the initial barrier. This would release GC% from its species-isolating role, thus allowing it to continue facilitating intraspecies genic divergence. Differences in intergenomic and intragenomic GC% are here considered separately. However, species with intragenomic isochore differentiation can themselves further differentiate into new species. In this case, a further layer of intergenomic GC% differentiation would be imposed upon a previous intragenomic differentiation. Again, when a sufficient degree of reproductive isolation had been achieved this initial barrier between species would usually be replaced by other barriers, thus leaving GC% free again to continue differentiating in response to intragenomic demands. By what mechanism would differences in GC% impede recombination? One model for the homology search that preceeds recombination requires initial "kissing" interactions between stem-loop structures extruded from duplex DNA [14, 16]. It has been shown elsewhere that such extrusion, and the required uniformity in the stem-loop pattern necessary for a successful paranemic homology search, would be critically affected by small differences in GC% [10]. |
|
Recombination can homogenize diverging genes within a genome or diverging genomes within a phylogenetic group, thus impairing evolutionary advance. The proposal that regions of relatively uniform GC% act as recombinational isolators is now much stronger than when advanced in 1968 [24]. The "gene homostabilization propensity" regions of Wada et al. [27, 30] are "microisochores." The classical isochores of Bernardi et al. are "macroisochores." As long suspected [5, 29], these small and large regions of relatively uniform GC% are likely to have the same function. Recombinational isolation may be a solution not only to the old problem of how a gene duplicate is initially sustained within a genome, but also to the even older problem how a genome duplicate is initially sustained within a phylogenetic group (e.g. the origin of species; [11, 12]). |
Acknowledgements
|
I thank J. R. Mortimer for Perl programs that extract base compositions from codon usage tables. Queen's University hosts my web-pages where full text versions of some of the cited references may be found. |
![]()
References
[1]
Bellgard,
M., Schibeci, D., Trifonov, E. and Gojobori, T. J., Early
detection of G + C differences in bacterial species inferred from the
comparative analysis of the two completely sequenced Helicobacter pylori
strains, J. Mol.
Evol.
53 (2001) pp. 465-468.
[2]
Bernardi,
G., Misunderstandings about isochores. Part 1, Gene 276 (2001) pp. 3-13.
[3]
Bibb,
M. J., Findlay, P. R. and Johnson, M. W., The
relationship between base composition and codon usage in bacterial genes and its
use for the simple and reliable identification of protein-coding sequences, Gene 30 (1984) pp.
157-166.
[4]
Dawkins,
R., The Extended Phenotype (W. H. Freeman, Oxford, 1982) pp. 84-89.
[5]
D'Onofrio,
G. and Bernardi, G., A universal compositional
correlation among codon positions, Gene 110 (1992) pp. 81-88.
[6]
Filipski,
J., Thiery, J. P. and Bernardi, G., An analysis of the bovine genome by Cs2SO4-Ag+
density gradient centrifugation, J. Mol. Biol. 80 (1973)
pp.177-197.
[7]
Forsdyke,
D. R., Percentage G + C determines frequencies of
complementary trinucleotide pairs: implications for speciation, Proc.
Can. Fed. Biol. Socs.
37 (1994) pp. 152.
[8]
Forsdyke,
D. R., Relative
roles of primary sequence and (G + C)% in determining the hierarchy of
frequencies of complementary trinucleotide pairs in DNAs of different species, J. Mol. Evol. 41 (1995) pp. 573-581.
[9]
Forsdyke,
D. R., Different biological species "broadcast" their
DNAs at different (G+C)% "wavelengths," J. Theor. Biol. 178
(1996) pp. 405-417.
[10]
Forsdyke,
D. R., An alternative way of thinking about stem-loops in
DNA. A case study of the G0S2 gene, J. Theor. Biol.
192 (1998) pp. 489-504.
[11] Forsdyke,
D. R., The Origin of Species, Revisited (McGill-Queen's University Press,
Montreal, 2001).
[12] Forsdyke,
D. R., William Bateson, Richard Goldschmidt, and non-genic
modes of speciation, J.
Biol. Sys. 11 (2003) pp. 341-350.
[13] Forsdyke,
D. R. and Mortimer, J. R., Chargaff's legacy, Gene
261 (2000)
pp. 127-137.
[14] Hawley,
R. S. and Arbel, T., Yeast genetics and the fall of the classical view of
meiosis, Cell 72 (1993) pp. 301-303.
[15] Iwase,
M., Satta, Y., Hirai, Y., Hirai, H. Imai, H. and Takahata, N. The
amelogenin loci span an ancient pseudoautosomal boundary in diverse mammalian
species, Proc. Natl. Acad. Sci. USA 100 (2003) pp. 5258-5263.
[16] Kleckner,
N., Interactions between and along chromosomes during meiosis. Harvey Lect.
91 (1997) pp. 21-45.
[17] Li,
W., Delineating
relative homogenous G + C domains in DNA sequences, Gene 276 (2001)
pp. 57-72.
[18] Matsuo,
K., Clay, O., Kunzler, P., Georgiev, O., Urbanek, P. and Schaffner, W., Short
introns interrupting the Oct-2 POU domain may prevent recombination between the
POU family genes without interfering with potential POU domain 'shuffling'
in evolution, Biol.
Chem. Hoppe-Seyler
375 (1994) pp. 675-683.
[19] Montoya-Burgos,
J. I., Boursot, P. and Galtier, N., Recombination
explains isochores in mammalian genomes, Trends. Genet.
19 (2003) pp. 128-130.
[20] Moore,
R. C. and Purugganan, M. D., The early stages of duplicate
gene evolution, Proc. Natl.
Acad. Sci. USA
100 (2003) pp. 15682-15687.
[21] Mortimer,
J. R. and Forsdyke, D. R., Comparison of responses by
bacteriophage and bacteria to pressures on the base composition of open reading
frames, Appl. Bioinform. 2
(2003) pp. 47-62.
[22] Muto,
A. and Osawa, S., The guanine and cytosine content of
genomic DNA and bacterial evolution.
Proc. Natl. Acad. Sci. USA 84 (1987) pp. 166-169.
[23] Nomura,
M., Sor, F., Yamagishi, M. and Lawson, M., Heterogeneity
of GC content within a single bacterial genome and its implications for
evolution, Cold Spring Harb. Symp.
Quant. Biol. 52 (1987) pp. 658-663.
[24] Skalka,
A., Burgi, E. and Hershey, A. D., Segmental distribution of
nucleotides in the DNA of bacteriophage lambda, J. Mol. Biol.
34 (1968) pp. 1-16.
[25] Suyama,
A. and Wada, A., Correlation between thermal stability
maps and genetic maps of double-stranded DNAs. J. Theor. Biol. 105 (1983) pp. 133-145.
[26] Vizard,
D.L. and Ansevin, A. T., High resolution thermal
denaturation of DNA: thermalites of bacteriophage DNA, Biochemistry
15 (1976) pp. 741-750.
[27] Wada,
A. and Suyama, A., Third letters in codons counterbalance
the (G + C) content of their first and second letters, FEBS Lett. 188 (1985) pp. 291-294.
[28] Wada,
A. and Suyama, A., Local stability of DNA and RNA
secondary structure and its relation to biological functions, Prog. Biophys. Mol. Biol.
47 (1986) pp. 113-157.
[29] Wada,
A., Suyama, A. and Hanai, R., Phenomenological theory of GC/AT
pressure on DNA base composition, J.
Mol. Evol.
32 (1991) pp. 374-378.
[30] Wada,
A., Tachibana, H., Gotoh, O. and Takanami, M., Long range
homogeneity of physical stability in double-stranded DNA, Nature
263 (1976) pp. 439-440.
[31] Xue,
H. Y. and Forsdyke, D. R., Low complexity segments in Plasmodium
falciparum are primarily nucleic acid level adaptations, Mol. Biochem.
Parasitol. 128 (2003) pp. 21-32.
[32] Zhang,
Z., Inomata, N., Ohba, T., Cariou, M-L. and Yamazaki, T., Codon
bias differentiates between duplicated Amylase loci following gene
duplication in Drosophila.
Genetics 161 (2002) pp. 1187-1196.
![]()
Go to Bioinformatics Index Click Here
Go to Home Page Click Here
Abstracts: Toronto 2003, Boston 2004 Click Here
![]()
This page was established in July 2004 and was last edited 08 Nov 2020 by Donald Forsdyke.