| Correlation of Chi orientation with transcription indicates a fundamental relationship between recombination and transcription |  |
| Correlation of Chi orientation with transcription indicates a fundamental relationship between recombination and transcription | |
Keywords: Base composition; Recombination islands; Chargaff's second parity rule; Szybalski's transcription direction rule.
S. J. Bell, Y. C. Chow, J. Y. K. Ho, and D. R. Forsdyke
Abbreviations: aa, aminoacid(s); Chi, Cross-over hot-spot instigator; kb, kilobase(s) or 1000 bp; ORF, open reading frame.
2.1. Location of sequence motifs
2.2. Chargaff difference analysis
3.1. Orientation of Chi and Chi-like sequences correlates best with transcription
3.2. GT-rich "recombination islands" in E. coli
3.3. GT-rich "recombination islands" in H. influenzae
3.4. Chargaff difference analysis of transcription direction
3.5. Comparison of natural and shuffled sequence
[Proceed directly to Lao & Forsdyke 2000]
| Cross-over hotspot instigator (Chi) sequences (5'-GCTGGTGG-3') are abundant, strand-specific, sequences, which locally increase recombination in E. coli. Located within G-rich "recombination islands", Chi orientations correlate with the orientations both of DNA replication and of transcription. Consistent with evidence from eukaryotic systems for a fundamental relationship between recombination and transcription, we find for Escherichia coli Chi sequences, and for Haemophilus influenzae Chi-like sequences, that orientations correlate better with transcription than with replication. Complying with Szybalski's transcription direction rule, open reading frames in these prokaryotes have purine-rich mRNA synonymous DNA strands. Hence, the G-richness of "recombination islands" may reflect their correspondence with "transcriptional islands" (genes). Comparison of a natural with the corresponding shuffled sequence, indicates a base order-dependent island unit of approximately 1 kb. |
|
Homologous chromosome pairing precedes meiotic recombination and may initiate, without strand breakage, by way of "kissing" interactions between the loops of extruded stem-loop structures (Kleckner, 1996; Forsdyke, 1996; 1998). A fundamental relationship between homologous chromosome pairing and transcription is suggested by studies in various eukaryotic systems (Stewart and Roeder, 1989; Tartof and Henikoff, 1991; Wu, 1993; Cook, 1997; Nicolas, 1998; Kodadek, 1998). This raises the question of a similar relationship in prokaryotic systems. A common feature of transcription and recombination is that both are orientated with respect to DNA. Recombination frequency in E. coli is greatly increased upstream (on the 5' side) of recombination "cross-over hot spot instigator" (Chi) sequences (Smith, 1997); these occupy GT-rich "islands" of a size (0.8 kb) approximating that of an average ORF (Tracy et al., 1997b). Chi is inactivated when inverted without change of location, showing that location in a strand is not arbitrary (Faulds et al., 1979). Chi recombination hot spots relate only to the sequence of one strand (5'-GCTGGTGG-3'), so occurrence of the complementary motif in a strand (5'-CCACCAGC-3'), means that Chi is in the opposite strand and influences recombination downstream of the complementary motif (on its 3' side). Much of the E. coli chromosome consists of ORFs which are transcribed either to the left or to the right of the promoter. An ORF has a mRNA-synonymous strand and a template strand. When transcription is to the right, the "top" strand is the mRNA-synonymous strand. When transcription to the left, the "bottom" strand is the mRNA-synonymous strand. Examination of a 227 kb section of the E. coli genome showed that Chi sequences, while abundant in both strands, occur preferentially in mRNA-synonymous strands (Burland et al., 1993). This could not be explained by asymmetrical distribution of potential codons contained in the Chi sequence, and supported a relationship between transcription and recombination. However, E. coli Chi sequences also occur preferentially in the leading strand of the replication fork (Blattner et al., 1997), indicating a relationship between Chi sequence orientation and the direction of DNA replication. Since replication direction sometimes correlates with transcription direction (Brewer, 1988), Chi sequence orientation might correlate with transcription direction as a consequence of the correlation with replication. A statistical analysis suggested the two correlations were independent (Burland et al., 1993). Chi-like sequences occur in a variety of organisms (Smith, 1997). Recently, replication direction was found to correlate poorly with the orientation of functional Chi-like sequences in Haemophilus influenzae (Sourice et al., 1998). This provided an opportunity to examine Chi orientation under conditions where the influence of replication was likely to be decreased. We have found for H. influenzae that there is a strong correlation between the orientations of Chi-like sequences and of transcription. We note that for E. coli there is a closer correlation of Chi sequence orientation with transcription direction than with replication direction. The G-richness of "recombination islands" (Tracy et al., 1997b) may reflect the fact that in these organisms the islands correspond to "transcription islands" which, according to Szybalski's transcription direction rule (Szybalski et al., 1966; Smithies et al., 1981; Forsdyke and Bell, 1997; Dang et al., 1998) are likely to have G-rich mRNA-synonymous strands. |
2.1. Location of sequence motifs
The Findpatterns program of the Genetics Computing Group, Madison, Wisconsin, was used to locate sequence motifs in the first 350 kb of H. influenzae Rd (bases 1 to 350,000; GenBank accession number L42023; Fleischmann et al., 1995), and the first 200 kb of E. coli (GenBank accession number U00096; Blattner et al., 1997). Motifs were counted in the "top" strand as designated in the GenBank record; to locate Chi sequences in the bottom strand, we sought the complement (5'-CCACCAGC-3') in the top strand. The significance of the distribution of motifs in three sectors (leftward-transcribing, rightward-transcribing, and non-ORF), relative to the probabilities of random localization (derived from the number of bases in each sector), was determined using the chi-square test (3 x 2 contingency table; Meyer and Kreuger, 1994). The significance of the ratios of occurrence in the mRNA-synonymous strand of DNA relative to occurrence in the mRNA-template strand was determined using a 2 x 2 contingency table, the probabilities of random localizations both being 0.5.
2.2. Chargaff difference analysis
When analysing the E. coli genome, Blattner et al. (1997) counting the members of a Watson-Crick base pair (C and G) in a moving 10 kb window, subtracted the number of Cs from the number of Gs, and then divided by their sum ("skew analysis"). This measure of compositional asymmetry can be related to replication direction (Smithies et al., 1981; Lobry, 1996), as can cumulative measures of base composition (Freeman et al., 1998). However, in E. coli the average ORF is only 317 aa, meaning that the average gene should correspond to about 1 kb of DNA. A 10 kb window might encompass 10 genes, some transcribing to the right and some to the left. Thus, a 10 kb window might not detect transcription direction-related compositional biases.
Parity between the members of each Watson-Crick base pair applies both to duplex DNA (Chargaff, 1951), and, to a close approximation, to single stranded DNA (Chargaff, 1979). Deviations from parity in the case of single stranded DNA ("Chargaff differences") were determined from the base composition of successive 1 kb windows which were moved along sequences in steps of 0.1 kb, as described by Dang et al. (1998). Chargaff differences were calculated as (A-T)/W and (C-G)/S and expressed as percentages. Here, A, T, C, and G refer to the number of the corresponding base in a window. W is the sum of the W bases (A+T) and S is the sum of the S bases (C+G). The significance of departures from parity was evaluated by Student's t-test.
To determine the window size at which Chargaff difference values were likely to be most biologically significant, the 200 kb natural E. coli segment was compared with the corresponding shuffled segment (base order randomized, but no change in overall base composition). Windows of varying size were moved along sequences in steps of 0.1 kb, and base compositions were determined in each window. Absolute Chargaff differences for the W and S bases were calculated as
dW/W and dS/S, respectively, and expressed as percentages. dW is the absolute value of the difference between the number of W bases (dW=|A-T|). dS is the absolute value of the difference between the number of S bases (dS=|C-G|; Dang et al., 1998).3.1. Orientation of chi and chi-like sequences correlates best with transcription
The first 350 kb of the 1,830 kb genome of H. influenzae was examined. Of this, 43.8% corresponds to leftward-transcribing ORFs, 37.9% corresponds to rightward-transcribing ORFs, and 18.3% corresponds to non-ORF DNA.
| . | Number of motifsa | . | Strand ratios | ||||
| ORFs | Non |
Transcription Synonoymous/ templatec |
Replication Leading/ laggingd |
||||
| Organism | Motif (Chi or Chi-like) |
Leftward | Rightward | Asym- metry (P)b |
|||
| H. influenzae | GNTGGTGG | 3 | 11 | 0 | 0.006 | 4.9 (44:9) P<0.00001 |
3.1 (1.6) |
| CCACCANC | 33 | 6 | 5 | 0.001 | . | . | |
| E. coli | GCTGGTGG | 7 | 36 | 1 | 0.001 | 5.5 (44:8) P<0.00001 |
4.9 (3.1) |
| CCACCAGC | 8 | 1 | 0 | 0.006 | . | . | |
| H. influenzae | AAGTGCGGTMe | 16 | 33 | 25 | 0.0006 | 1.4 (56:37) P=0.05 |
0.9 |
| KACCGCACTT | 23 | 21 | 23 | 0.002 | . | . | |
| a Motifs were counted in the "top" strand as
designated in the GenBank records (the first 350 kb of H. influenzae Rd, accession
number L42023, and the first 200 kb of E. coli, accession number U00096. b The significance of the asymmetrical distribution in the three sectors (leftward, rightward, non-ORF), relative to the probabilities of random localization (derived from the number of bases in each sector).c The ratio of occurrence in the mRNA-synonymous strand of DNA relative to occurrence in the mRNA-template strand. The probabilities that the departures from equal distributions (i.e. ratio 1.0) are not significant are in parenthesis.d The ratios of occurrences of Chi in the leading strand of DNA replication, relative to occurrences in the lagging strand, are shown for the segments examined, and for the whole genomes (data from Sourice et al., 1998 in parentheses).e Uptake signal sequence (Smith et al., 1985). |
|||||||
Table 1 shows that the Chi-like motif 5'-GNTGGTGG-3' appears 14 times in the "top" strand, and 44 times in the "bottom" strand (its complement being recorded in the top strand as 5'-CCACCANC-3'). At this location in the genome, the top strand corresponds to the lagging strand of DNA replication, and the leading/lagging strand distribution ratio (3.1) is higher than the genome average of 1.6 (Sourice et al., 1998). Of the total number of Chi occurrences (14+44=58), only 5 are non-ORF.
Combining data from both strands, the ratio of occurrence in the mRNA synonymous strand relative to occurrence in the mRNA template strand (4.9), is greater than the ratio of occurrence in the leading strand of DNA replication, relative to occurrence in the lagging strand. Similar results were obtained for the distribution of Chi sequences in the first 200 kb of the 4,639 kb E. coli genome (Blattner et al., 1997). Here the ratio of occurrence in the mRNA-synonymous strand relative to the mRNA template strand is 5.5, which is again higher than the ratio for the occurrences in the leading strand relative to the lagging strand (4.9).
In contrast, a sequence containing the abundant species-specific motif governing uptake of exogenous DNA by H. influenzae (5'-AAGTGCGGT-3'; Smith et al., 1995; Sourice et al., 1998) shows a strong preference for non-ORF DNA; the remaining ORF-associated motifs show a minimal, but significant, bias for the mRNA-synonymous strand.
3.2. GT-rich "recombination islands" in E. coli
In E. coli the Chi sequence occurs within approximately 1 kb GT-rich "recombination islands", where the purine base G is particularly abundant (Tracy et al., 1997b). In these regions, for the S bases G>C, and for the W bases T>A. To examine this further, a 1 kb window was moved through the 200 kb segment in 0.1 kb steps, and the base composition of each window determined. For the classical Watson-Crick pairing bases, differences from parity ("Chargaff differences") were plotted against each other. In this way the relative distribution of bases between four segments (AG, AC, TG, TC) is readily visualized ("quadrant analysis"; Fig. 1).
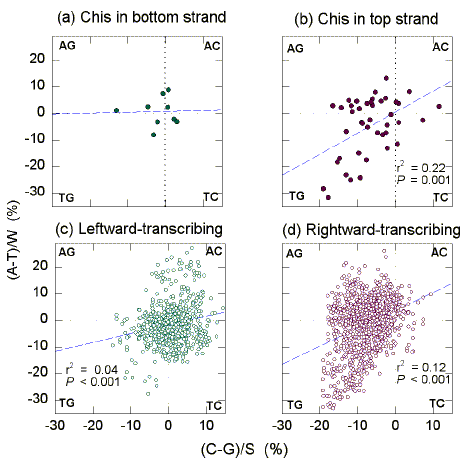 |
| Fig. 1. Quadrant analysis of Chargaff differences for the W bases and the S bases in 1 kb windows from the "top" strand of the first 200 kb of the E. coli genome. Each quadrant corresponds to windows enriched for two particular bases (indicated at the corners). (a) The 9 windows whose centres are closest to Chi sequences in the bottom strand. (b) The 44 windows whose centres are closest to Chi sequences in the top strand. (c) The 746 windows whose centres overlap leftward-transcribing ORFs. (d) The 1074 windows whose centres overlap rightward-transcribing ORFs. Dashed diagonals are the least-squares regression lines. Listed are the squares of the correlation coefficients (r2) and the probabilities (P) that slopes are not significantly different from zero. |
Chi sequences are distributed in the E. coli genome at approximately 5 kb intervals (Smith, 1997). Windows whose centres were closest to Chi sequences were selected. In the case of the 44 top strand Chi sequences in the 200 kb segment, the corresponding windows tended to be GT-rich (Fig. 1b), but there was no evidence of a trend in the case of the 9 bottom strand Chi sequences (Fig. 1a). For the top strand Chi sequences the average Chargaff difference for the W bases, calculated as (A-T)/W and expressed as a percentage, was -4.9 se 1.7 (i.e. T>A; Table 2). The corresponding value for the S bases, calculated as (C-G)/S and expressed as a percentage, was -6.9 se 1.0 (i.e. G>C). Chargaff differences values for the W bases and for the S bases were correlated (P=0.001; Fig. 1b). Thus, if a Chi window is enriched in G it also tends to be enriched in T. When plotted in a similar fashion, no significant correlations were noted for the 9 Chi sequences in the "bottom" strand (Fig. 1a).
| . | Chi window sub set | Total windows | |||
| Strand orientation | Transcriptional orientation | ||||
| Species | Chargaff difference (%) |
Bottom | Top | To left | To right |
| E. coli | (A-T)/W | -a | T>A -4.9 se 1.7b (P<0.01) |
T>A -1.5 se 0.3 (P<0.001) |
T>A -1.3 se 0.3 (P<0.001) |
| (C-G)/S | -a | G>C -6.9 se 1.0 (P<0.001) |
C>G 0.5 se 0.2 (P<0.01) |
G>C -7.6se 0.1 (P<0.001) |
|
| H. influenzae | (A-T)/W | A>T 1.3 se 1.2 (P>0.10) |
T>A -3.6 se 1.8 (P<0.10) |
A>T 0.3 se 0.2 (P>0.10) |
A>T 2.1se 0.2 (P<0.001) |
| (C-G)/S | C>G 10.0 se 1.3 (P<0.001) |
G>C -9.3 se 2.8 (P<0.01) |
C>G 10.2 se 0.2 (P<0.001) |
G>C -3.6 se 0.2 (P<0.001) |
|
|
aSince there were only 9 Chi sequences in the bottom strand of the 200 kb
segment studied ( see
Fig. 1a), departures from parity (Chargaff differences) are not shown. b Each data set refers to (i) the relative proportions of the relevant Watson-Crick base pair, (ii) the corresponding Chargaff difference value (se = the standard error of the mean), and (iii) the probability that the value is not significantly different from zero (Student's t-test). |
|||||
Since Chi sequences in the top strand tend to be located in rightward-transcribing ORFs, the base composition of the corresponding windows with Chi sequences at their centres might reflect the base composition of rightward-transcribing ORFs in general, which can be predicted from Szybalski's transcription direction rule (i.e. purine-rich; see Discussion and Dang et al., 1998). In this circumstance, windows containing top strand Chi sequences would be a subset of all rightward-transcribing windows, and would have characteristics of the latter. Indeed, rightward-transcribing ORFs generally tend to be GT-rich (Fig. 1d), and this applies particularly to the purine. The average Chargaff difference for the W bases was -1.3 se 0.3 (i.e. T>A; Table 2). The corresponding value for the S bases was -7.6 se 0.1 (i.e. G>C). For leftward-transcribing ORFs, Szybalski's rule predicts pyrimidine-richness. Indeed, the average Chargaff difference for the W bases was -1.5 se 0.3 (i.e. T>A), and the corresponding value for the S bases was 0.5 se 0.2 (i.e. C>G).
For all windows in the E. coli segment, Chargaff differences for the W bases correlate positively with Chargaff differences for the S bases (P<0.001; Figs. 1c, d). The slope of the line corresponding to the 44 top-strand Chi sequences (0.79; Fig. 1b) is not significantly different from the slope of the line for all rightward-transcribing ORFs (Fig. 1d; P=0.60), but is significantly different from the slope of the line for all leftward-transcribing ORFs (Fig. 1c; P=0.01). The slope of the plot for windows corresponding to rightward-transcribing ORFs (0.67; Fig. 1d), was greater than the slope for windows corresponding to leftward-transcribing ORFs (0.33; Fig. 1c; P<0.001). For this 200 kb segment there is an asymmetry; rightward-transcribing regions tend to be GT-rich, but this is not matched by an equal tendency of leftward-transcribing regions to be AC-rich.
3.3. GT-rich "recombination islands" in H. influenzae
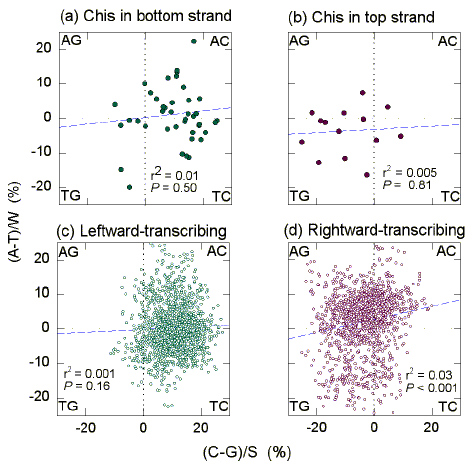 |
| Fig. 2 Quadrant analysis of Chargaff differences for the W bases and the S bases in 1 kb windows from the "top" strand of the first 350 kb of the H. influenzae genome. (a) The 44 windows whose centres are closest to Chi-like sequences in the bottom strand. (b) The 14 windows whose centres are closest to Chi-like sequences in the top strand. (c) The 1562 windows whose centres overlap leftward-transcribing ORFs. (d) The 1326 windows whose centres overlap rightward-transcribing ORFs. Other details are as in Fig. 1. |
Similar results were obtained in the case of the first 350 kb of the H. influenzae genome (Fig. 2; Table 2). Reflecting the base composition of windows corresponding to rightward transcribing ORFs (Fig. 2d), windows corresponding to Chi sequences in the top strand tend to be GT-rich (Fig. 2b). Reflecting the base composition of windows corresponding to leftward-transcribing ORFs (Fig. 2c), windows corresponding to Chi sequences in the bottom strand tend to be AC-rich (Fig. 2a), the latter referring to the composition of the top strand. This means that bottom strand Chi sequences tend to be embedded in a locally GT-rich strand like their top strand counterparts. For the top strand Chi sequences (Fig. 2b) the average Chargaff difference for the W bases was -3.6 se 1.8 (i.e. T>A; Table 2). The corresponding value for the S bases was -9.3 se 2.7 (i.e. G>C). For the bottom strand Chi sequences (Fig. 2a) the average Chargaff difference for the W bases was 1.3 se 1.2 (i.e. A>T). The corresponding value for the S bases was 10.0 se 1.3 (i.e. C>G). For all windows transcribed to the right (Fig. 2d) the average Chargaff difference for the W bases was 2.1 se 0.2 (i.e. A>T). The corresponding value for the S bases was -3.6 se 0.2 (i.e. G>C). For all windows transcribed to the left (Fig. 2c) the average Chargaff difference for the W bases was 0.3 se 0.2 (i.e. A>T). The corresponding value for the S bases was 10.2 se 0.2 (i.e. C>G).
3.4. Chargaff difference analysis of transcription direction
That Szybalski's transcription direction rule applies in local regions of DNA is evident from detailed studies of sequence features, and can be employed to assist the identification of functional ORFs (Dang et al., 1998). For example, Fig. 3 shows Chargaff differences for a 20 kb segment corresponding to the 100-120 kb region of the E. coli genome. If the line for the S bases (C and G) is followed from left to right (red symbols), it is seen that G>C in the case of the rightwardly-transcribed first half of the segment, where most of the genes are unambiguous.
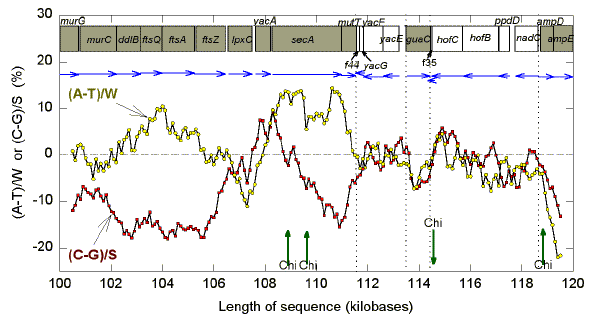 |
| Fig. 3. Variation of "Chargaff differences" ("G-C skew") along a 20 kb segment from E. coli K-12. A 1 kb window was moved in steps of 100 nt and base compositions were determined in each window. Chargaff differences for the S bases (C and G; red squares) are calculated as (C-G)/S (where S = C+G). Chargaff differences for the W bases (A and T; yellow circles) are calculated as (A-T)/W (where W=A+T). Data points are located at the center of each window. The proposed locations of ORFs are shown as boxes. Horizontal blue arrows indicate the transcription directions assigned to each ORF by Blattner et al. (1997), with rightward ORFs being represented by filled boxes. Vertical arrows indicate the positions of Chi sequences either in the top strand (upward pointing), or in the bottom strand (downward pointing). Vertical dotted lines correspond to regions where the transcription direction switches. Abbreviated gene names are as in Blattner et al. (1997). |
An exception is a small hypothetical ORF (yacA) in the lpxC-secA intergenic region, which would require an unusual start codon (GTG), and has C>G. The latter would argue against the ORF being functional. There is a hypothetical small leftward-transcribing ORF (f44; 44 amino acids) which also appears to be in conflict with Szybalski's rule (G>C instead of C>G), so also may not be functional. The leftward-transcribing group (yacG, yacF, and yacE) tends to conform (C
>G), as does the rightwardly transcribing guaC encoding GMP reductase (G>C). Genes encoding transport proteins (hofC, hofB) are leftward transcribing, and consistent with this, C>G. The gene encoding prepilin peptidase dependent protein D (ppdD) is unambiguously identified, but clearly does not conform to the rule. Hence, the rule is not invariant. The rightwardly transcribing genes encoding ampD and AmpE clearly conform (G>C).The positions of Chi sequences in the "top" strand (the mRNasynonymous strand when transcription is to the right) are marked by upward pointing arrows, and one in the "bottom" strand (the mRNA synonymous strand when transcription is to the left) is marked by a downward-pointing arrow. The top strand Chi sequences are in regions where transcription is to the right, and the bottom strand Chi sequence is in a region where transcription in to the left. The first two top strand Chi sequences are close to each other so that their "recombination islands" would slightly overlap. The region is purine-rich (A and G) rather than GT-rich. The third top strand Chi sequence is in a GT-rich region. The bottom strand Chi sequence would be expected to be located preferentially in a region where the top strand would be AC-rich, but this particular Chi locates to a region where Chargaff differences are small.
3.5. Comparison of natural and shuffled sequence
In Fig. 4 average absolute Chargaff differences are plotted against the size of sequence windows in the above 200 kb segment from E. coli. With windows of only 200 nt, high differences would be expected since there would be great statistical fluctuations when base "coins" are "tossed" no more than 200 times. Indeed, average absolute differences for both the W bases and the S bases are high when windows are 200 nt. However, values for the natural sequence exceed those of the shuffled natural sequence, implying an evolutionary selection pressure on base order favouring non-parity between the Watson-Crick base pairs.
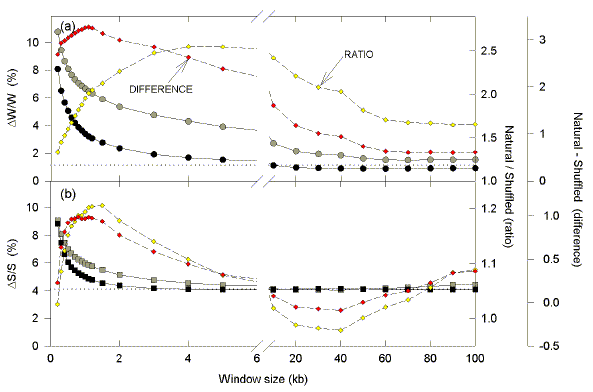 |
| Fig. 4. Variation of average Chargaff difference values with size of windows in E. coli. Windows of varying size were moved along the 200 kb segment in steps of 100 nucleotides, and base compositions were determined in each window. Average absolute Chargaff differences for each window size are plotted either as large grey symbols (natural sequence), or as large black symbols (shuffled sequence). Small yellow diamonds refer to the ratio of the se values (the average Chargaff difference for the natural sequence divided by the average Chargaff difference for the shuffled sequence). Small red diamonds refer to the difference between these values determined by subtraction. The horizontal dotted lines indicate Chargaff differences for the entire sequence (i.e. the largest possible window, of which there is only one copy). Thus, the total number of windows of a given size varies with sequence length. In a 100 kb sequence there will be 999 windows of 0.2 kb, and one window of 100 kb. |
With increasing window size, average Chargaff differences for both natural and shuffled sequences decrease exponentially to approach the value for the entire segment (horizontal dotted lines). Values for the natural and shuffled sequences were compared either as a ratio (open yellow diamonds), or by subtraction (filled red diamonds). The size of the window at which Chargaff differences for natural and shuffled sequences diverge maximally depends on the method used. In the case of the W bases the maximum divergence by ratio occurs with 4 kb windows, but the maximum divergence by subtraction is with 1.1 kb windows. In the case of the S bases the divergence by the ratio method is high with 1 kb windows, but reaches at maximum with 1.5 kb windows. By the difference method, the divergence reaches a maximum level at 0.6 kb which is sustained to 1.2 kb. Thus, the base order of the natural sequence has been under evolutionary constraint, and for E. coli the window size at which this is maximally evident is about 1 kb. This is consistent with a fundamental transcription/recombination "island" unit size of approx. 1 kb.
In 1951 Chargaff reported the famous equality of the Watson-Crick pairing-bases in duplex DNA (%A=%T, %C=%G; Chargaff's first parity rule), and in 1968 showed that, to a close approximation, this also applied to single strands of DNA (Chargaff's second parity rule; Rudner et al., 1968; Chargaff, 1979). In 1966 Szybalski et al. reported that in certain microorganisms RNA polymerase template strands contained pyrimidine clusters; hence, the corresponding mRNA synonymous DNA strands contained purine clusters.
Combining the observations of Chargaff, Szybalski and their coworkers, it appeared possible that when transcription was to the right there would be a compositional bias, a small deviation from the second parity rule, in favour of purines; similarly, when transcription was to the left the bias would favour pyrimidines (see the prescient Fig. 4 of Szybalski et al., 1966). Studies of Smithies et al. (1981) suggested that this might apply both to prokaryotes and eukaryotes, as has been confirmed (S. J. Bell and D. R. Forsdyke, [1999a,b; unpublished in 1998]; Forsdyke and Bell, 1997). This regularity, which we refer to as Szybalski's transcription direction rule, can assist the prediction of functional ORFs (Dang et al., 1998). In the case of low (C+G)% genomes, the purine predictor of rightward-transcription tends to be A, whereas in the case of average or high (C+G)% genomes, the purine predictor tends to be G. Assuming the examined segments to be representative, for both H. influenzae (C+G = 38%), and E. coli (C+G = 51%), the best predictor of rightward-transcription is G (Figs. 1-3; Table 2). However, for leftward-transcription, T is the best pyrimidine predictor for E. coli, and C is the best for H. influenzae.
Since contiguous ORFs may be transcribed in different directions, in one section of DNA one may encounter regions enriched in purines (rightward-transcription), and pyrimidines (leftward-transcription; Fig. 3). If the order of bases in the region were shuffled, the bias would be decreased, and Chargaff differences would be minimized. Thus, by comparing Chargaff differences in natural and shuffled sequences, one arrives at a window size (1 kb) which is most likely to be biologically informative (Fig. 4). Below this window size stochastic factors make correlations with ORFs difficult. Above this window size, purine-rich regions tend to cancel out pyrimidine-rich regions (S. J. Bell and D. R. Forsdyke [1999a,b; unpublished in 1998]; Dang et al., 1998). The critical window size approximates that of the average transcription unit. Remarkably, this corresponds closely to the size of Chi domains which Tracy et al. (1997b) refer to as "recombination islands".
Our studies show that a relationship between recombination and transcription is more evident than a relationship between recombination and replication (Table 1). Correlations between the orientations of Chi (or Chi-like) sequences, and transcription directions, support the case for a fundamental relationship between domains of recombination ("recombination islands"), and domains of transcription ("transcription islands"). Chargaff difference quadrant analysis further emphasizes the linkage between recombination and transcription (Figs. 1, 2). G-rich sequences bind preferentially to the E. coli RecA protein and its yeast homolog (Rad51; Tracy et al., 1997a). The G-richness of strand-specific Chi-domains seems to reflect the fact that they occupy mRNA-synonymous DNA strands which are generally purine-rich (Table 2). However, in the case of the AT-rich H. influenzae genome, windows with Chi-like sequences at their centres may be a distinct subset of purine-rich windows. This is particularly apparent in the case of rightward-transcribed ORFs where both A and G contribute to the general purine richness, whereas mainly G contributes to top strand windows with Chi-like sequences at their centres (Fig. 2; Table 2). A possible explanation for "purine-loading" of mRNA synonymous strands is considered elsewhere (Cristillo et al., 1998).
Acknowledgements
We thank J. T. Smith for statistical advice, J. H. Gerlach and A. D. Cristillo for assistance with computer configuration, and Queen's University for financial support.
References
Blattner, F. R. et al., 1997. The complete genome sequence of Escherichia coli K-12. Science 277, 1453-1474.
Brewer, B. J., 1988. When polymerases collide: replication and the transcriptional organization of the Escherichia coli chromosome.
Cell 53, 679-686.Burland, V., Plunkett, G., Daniels, D. L., Blattner, F. R., 1993. DNA sequence and analysis of 136 kilobases of the Escherichia coli genome: organizational symmetry around the origin of replication. Genomics 16, 551-561.
Chargaff, E., 1951. Structure and function of nucleic acids as cell constituents. Fed. Proc. 10, 654-659.
Chargaff, E., 1979. How genetics got a chemical education. Ann. N. Y. Acad. Sci. 325, 345-360.
Cook, P. R., 1997. The transcriptional basis of chromosome pairing. J. Cell Sci. 110, 1033-1040.
Cristillo, A. D., Lillicrap, T. P., Forsdyke, D. R., 1998. Purine loading of EBNA-1 mRNA avoids sense-antisense "collisions". FaseB. J. 12, A1453 (abstract no. 828).
Dang, K. D., Dutt, P. B., Forsdyke, D. R., 1998. Chargaff difference analysis of the bithorax complex of Drosophila melanogaster. Biochem. Cell Biol. 76, 129-137.
Faulds, D., Dower, N., Stahl, M. M., Stahl, F. W., 1979. Orientation-dependent recombination hotspot activity in bacteriophage l . J. Mol. Biol. 131, 681-695.
Fleischmann, R. D. et al., 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496-512.
Forsdyke, D. R., 1996. Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths". J. Theor. Biol. 178, 405-417.
Forsdyke, D. R. 1998. An alternative way of thinking about stem-loops in DNA. A case study of the G0S2 gene. J. Theor. Biol. 192, 489-504.
Forsdyke, D. R., Bell, S. J., 1997. Deviations from Chargaff's second rule correlate with direction of transcription and genome structure. Proc. Can. Fed. Biol. Socs. 40, 87 (abstract no. 260).
Freeman, J. M., Plasterer, T. N., Smith, T. F., Mohr, S. C., 1998. Patterns of genome organization in bacteria. Science 279, 1827.
Kleckner, N., 1996. Meiosis: how could it work? Proc. Natl. Acad. Sci. USA 93, 8167- 8174.
Kodadek, T., 1998. Mechanistic parallels between DNA replication, recombination and transcription. Trends Biochem. Sci. 23, 79-83.
Lobry, J. R., 1996. Origin or replication of Mycoplasma genitalium. Science 272, 745-746.
Meyer, R. K., Krueger, D. D., 1994. Minitab Computer Supplement. Macmillan College Publishing, New York, pp. 228-229.
Nicolas, A., 1998. Relationship between transcription and initiation of meiotic recombination: towards chromatin accessibility. Proc. Natl. Acad. Sci. USA 95, 87-89.
Rudner, R, Karkas, J. D., Chargaff, E., 1968. Separation of B. subtilis DNA into complementary strands, III. Direct analysis. Proc. Natl. Acad. Sci. USA 60, 921-922.
Smith, G. R., 1997. Chi sites and their con sequences. In: Bruijn, F.J. de, Lipski, J.R., Weinstock, G. M. (Eds.), Bacterial Genomics. Thomson Publishing, New York, pp. 49-66.
Smith, H. O., Tomb, J. -F., Dougherty, B, A., Fleischmann, R. D., Venter, J. C., 1995. Frequency and distribution of DNA uptake signal sequences in the Haemophilus influenzae Rd genome. Science 269, 538-540.
Smithies, O., Engels, W. R., Devereux, J. R., Slightom, J. L., Chen, S-h., 1981. Base substitutions, length differences and DNA strand asymmetries in the human G
g and Ag fetal globin gene region. Cell 26, 345-353.Sourice, S., Biaudet, V., El Karoui, M., Ehrlich, S. D., Gruss, A., 1998. Identification of the Chi site of Haemophilus influenzae as several sequences related to the Escherichia coli Chi site. Mol. Microbiol. 27, 1021-1029.
Stewart, S. E., Roeder, G. S., 1989. Transcription by RNA polymerase I stimulates mitotic recombination in Saccharomyces cerevisiae. Mol. Cell. Biol. 9, 3464-3472.
Szybalski, W., Kubinski, H., Sheldrick, P., 1966. Pyrimidine clusters on the transcribing strand of DNA and their possible role in the initiation of RNA synthesis. Cold Spring. Harb. Symp. Quant. Biol. 31, 123-127.
Tartof, K. D., Henikoff, S., 1991. Trans-sensing effects from Drosophila to humans. Cell 65, 201-203.
Tracy, R. B., Baumohl, J. K., Kowalczykowski, S. C., 1997a. The preference for GT-rich DNA by the yeast Rad51 protein defines a set of universal pairing sequences. Genes Devel. 11, 3423-3431.
Tracy, R. B., Chedin, F., Kowalczykowski, S. C., 1997b. The recombination hot spot Chi is embedded within islands of preferred DNA pairing sequences in the E. coli genome. Cell 90, 205-205.
Wu, C, -t., 1993. Transvection, nuclear structure, and chromatin proteins. J. Cell Biol. 120, 587-590.
Go to: Chi sequences in Recombination/Transcription "Islands" (Click Here)
Return to: Bioinformatics Index (Click Here)
Return to: Evolution Index (Click Here)
Return to: HomePage (Click Here)
Last edited on 09 November 2020 by D. R. Forsdyke