|
Crossover Hotspot Instigator (Chi) sequences in E. coli occupy distinct recombination/transcription islands P. J. Lao, D. R. Forsdyke Gene (2000) 243, 47-57 Copyright
permission from Elsevier pending.
|
|
|
Crossover Hotspot Instigator (Chi) sequences in E. coli occupy distinct recombination/transcription islands P. J. Lao, D. R. Forsdyke Gene (2000) 243, 47-57 Copyright
permission from Elsevier pending.
|
|
2.1. Genomic sequences
2.2. Location of Chi sequences
2.3. Chargaff difference analysis3.1. Chi frequency exceeds that expected from (C+G)%
3.2. Orientation of Chi correlates with transcription
3.3. Chi sequences in GT-rich "islands" in E. coli
3.4. No Chi sequences in GT-rich "islands" in M. jannischii
3.5. Chi distribution relative to replication and transcription directions4.1. Mechanism of homologous recombination
4.2. Evolution of recombination
4.3. Codon usage "explains"?
4.4. Chi sequences in other bacteria
![]()
| Abstract Cross-over hot-spot instigator (Chi)sequences (5'-GCTGGTGG-3') are orientation-dependent, strand-specific sequences implicated in RecA-mediated DNA recombination. In E. coli and H. influenzae Chi and Chi-like sequences preferentially locate to approx. 1 kb recombination "islands" in the mRNA-synonymous strands of open reading frames (ORFs). Since mRNA-synonymous strands follow Szybalski's transcription direction rule in being G-rich, and the average ORF is about 1 kb, then, on this basis alone, Chi sequences are seen to reside in 1 kb G-rich "islands". However, RecA preferentially binds GT-rich sequences, suggesting that genomic context might potentiate Chi action. Consist with this, we report for E. coli that 1 kb sequence windows with Chi near their centres are a distinct subset of total 1 kb windows, the mRNA-synonymous strands being preferentially enriched in both G and T. Chi function might be particularly important for bacteria which survive high temperature and radiation. These often exist in habitats where recombination with E. coli DNA would be unlikely, so canonical Chi sequences might not confer a selective disadvantage in this respect. In general, Chi sequences are not more frequent in thermophilic bacteria and D. radiodurans, than in E. coli and other mesophilic bacteria. Only two of five thermophilic bacteria examined showed preferential location of Chi sequences to mRNA-synonymous strands. In the thermophile M. jannaschii, windows containing the canonical Chi sequence do not form a distinct subset. We suggest that in thermophilic bacteria and D. radiodurans the Chi function may be achieved by sequences which differ from the canonical Chi sequence, or the number of these sequences is sufficient, or the Chi function is unnecessary. |
![]()
| Cross-over hot-spot instigator (Chi) sequences (5'-GCTGGTGG-3') are abundant, strand-specific, sequences, which locally increase recombination in Escherichia coli. Noting regularities in surrounding sequences, Tracy et al. (1997b) suggested that canonical 8-base Chi sequences in E. coli usually exist within approximately one kb GT-rich "recombination islands". A correlation of Chi orientation with transcription direction (Burland et al., 1993) suggested that the islands might also be "transcription islands", implying a relationship between transcription and recombination (Bell et al., 1998; Bell and Forsdyke, 1999a,b). Since in E. coli open reading frames (ORFs) form the main component of transcripts, the islands could also be considered "translational islands" (Biaudet et al., 1998; Colbert et al., 1998). Mutations creating or inactivating Chi activity occur only in Chi, not in surrounding DNA (Smith, 1997), and insertion of the Chi octomer into DNA enhances recombination in all cases reported (Dixon and Kowalczykowski, 1991; Dabert et al. 1992; Kuzminov et al., 1994). These observations imply that the octomer alone is sufficient for activity. However, recombination hotspots also occur in eukaryotes, where they may be influenced by chromosomal context (Ponticelli and Smith, 1992). Noting that RecA, a key enzyme influenced by Chi, binds preferentially to GT-rich DNA, it was suggested that the genomic context of Chi might potentiate its effectiveness (Tracy et al., 1997a,b). If this were so, then it is possible that one kb sequence "windows" with Chi sequences at their centres might form a distinct subset of the corresponding total set of one kb windows. This characteristic, together with a preference for mRNA-synonymous regions of DNA, could also indicate whether Chi or Chi-like sequences in other genomes are likely to be functional. Absence of the appropriate Chi function "password" targets an exogenous DNA for exonucleolytic degradation by the host cell RecBCD homolog (Kuzminov et al., 1994), thus militating against interspecies recombination, while permitting intraspecies recombination. Disruption of such interspecies barriers facilitates recombination (Rayssiguier et al., 1989). Just as a general (C+G)% similarity between the DNAs of members of a species may be necessary for the initiation of homologous recombination (Forsdyke, 1996), so exogenous DNA must have sufficient Chi sequence similarity with the DNA of host bacteria. However, the selection pressure promoting differentiation of sequences mediating the Chi function would be unlikely to be sustained between species occupying different ecological niches. This might explain why E. coli and H. influenzae have similar Chi sequences (Sourice et al., 1998). To examine these issues, we here present a study of
Since Chi-dependent recombination could be involved in the repair of DNA strand breaks, the study included bacteria which both occupy ecological niches where recombination with E. coli would be unlikely (hence no selection pressure against the canonical Chi sequence), and survive under conditions where DNA strand breakage is increased (high temperature and radiation). |
|
Sequence information refers to the "top" strand as designated in the GenBank record. Unedited segments of the Deinococcus radiodurans genome were obtained in 1997 from the Institute for Genome Research, Rockville, MD. Five of the longest segments, which had been designated gdr_5 (77466 nt), gdr_19 (68159 nt), grd_23 (50515 nt), gdr_31 (60954 nt), and gdr_34 (61120 nt), were presumed to originate from disparate parts of the genome, and hence were considered likely to be representative of the genome. Putative ORFs were determined using the Glimmer program. 2.2. Location of Chi sequences Chi sequences were located using the Findpatterns program of the Genetics Computing Group, Madison, WI. Since it is established experimentally that H. influenzae uses both the canonical and degenerate "Chi-like" sequences (Sourice et al., 1998), we sought such sequences (GNTGGTGG) and their top strand complement (CCACCANC) in this organism. In all other cases, we sought the canonical Chi sequence in the top strand, with Chis in the bottom strand being detected as the complement (CCACCAGC) in the top strand. Chi sequences were classified according to their location in leftward or rightward-transcribed ORFs. For E. coli and H. influenzae locations in non-protein-encoding genes (e.g. ribosomal RNA) were also sought in the examined segments, but were found only in H. influenzae. For each organism, the frequency of Chi sequences (Chi sequences/kb) expected if base order were random was calculated from the base composition of the combined genomic segments under study; this was the product of each base's statistical chance of occurrence in each Chi octomer, multiplied by the number of possible independent octomers in 1 kb (993). Thus, if P(A)=P(T)=0.2 and P(C)=P(G)=0.3, then the expected frequency for GCTGGTGG and CCACCAGC in a sequence is [[(0.3)6(0.2)2]+[(0.3)6(0.2)2]] x 993 = 0.058. 2.3. Chargaff difference analysis Chargaff's first parity rule for duplex DNA (%A = %T; %C = %G) applies, to a close approximation, to single stranded DNA (Chargaff's second parity rule). Deviations from parity are referred to as "Chargaff differences", which often depend on the "purine loading" of mRNA synonymous strands (providing an explanation for Szybalski's transcription direction rule; Bell and Forsdyke, 1999b). The base-composition of successive 1 kb windows, moved in steps of 0.1 kb, was assessed as described by Dang et al. (1998). Chargaff differences were calculated as (A-T)/W and (C-G)/S and expressed as percentages. Here, A, T, C and G refer to the frequency of the corresponding base in a window. The direction of subtraction (A-T or T-A) is determined alphabetically. W is the sum of the W base frequencies (A+T) and S is the sum of the S base frequencies (C+G). A window of 1 kb was chosen as the size which optimizes the difference between the Chargaff differences of a natural and the corresponding shuffled sequence (Bell and Forsdyke, 1999a). A 1 kb window selected by virtue of containing a Chi octomer near its centre would receive a contribution to the Chargaff difference from the octomer by virtue of the excess of Gs over Cs (4) and of Ts over As (1). Thus, for an organism of 50% (C+G), the G excess would contribute [4/500] x 100 = 0.8% to the Chargaff difference percentage value. Differences of this order would only marginally have affected results due to compositional differences in the entire 1 kb "island" surrounding a Chi sequence. |
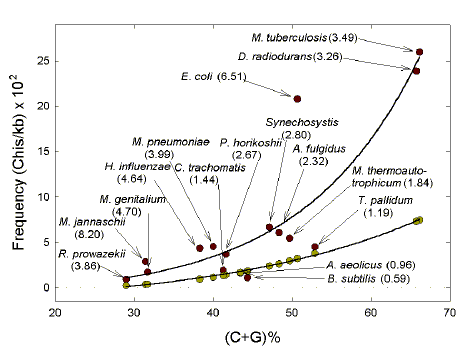
| Due to base composition alone, the frequency of the G-rich Chi sequence would be expected to increase as genome (C+G)% increases. However, whatever the (C+G)%, in most bacteria examined the observed frequency of the canonical Chi sequence is greater than expected (Fig. 1; exceptions A. aeolicus, B. subtilis, T. pallidum). The points corresponding to observed and expected Chi frequencies fit simple exponential curves, with the prominent exception of E. coli. However, the observed/expected frequency ratio for E. coli (6.51) is of the same order as that of most other bacteria, and is slightly less than that of the thermophile M. jannaschii (8.20). For this organism with four other thermophiles the mean ratio is 3.20 se 1.28; the mean ratio of the eleven mesophilic bacteria studied is 3.32 se 0.52. |
|
|
| Fig. 1.
Higher than expected frequency of canonical Chi sequences in many bacterial genomes. Points for observed frequency (red circles) and for the frequency expected if base order were random (green circles) were reiteratively fitted to corresponding least-squares curves to arrive at the formulae: Observed frequency = -1.049 + 0.2917e0.0681 x (C+G)% (r2 = 0.749; P<0.0001); Expected frequency = -2.118 + 0.6867e0.04 x (C+G)% (r2 = 0.997; P<0.0001). Organisms corresponding to each point are identified, with ratios of observed to expected frequencies in parentheses. segments examined are as in Table 1. The standard error of estimation for the curve fitted to the observed frequencies was 4.17, which gives a standardized residual for the E. coli outlier of >3.0 (indicating that its outlier status is statistically highly significant). For H. influenzae only data for canonical Chi sequences occurrence are shown. |
3.2. Orientation of Chi correlates with transcription
In the case of H. influenzae, Chi and Chi-like sequences are likely to be functional (Sourice et al., 1998). This was supported by the demonstration that, as in E. coli, the Chi and Chi-like sequences of H. influenzae tend to locate to ORFs, where they are found in mRNA-synonymous strands rather than in template-strands (Bell et al., 1998). Accordingly, we examined whether this preference for mRNA-synonymous strands also applied to the Chi sequences of other bacteria (Table 1).
| Bacteriuma | (C+G) % | Genome length (Mb) | segment examined | DNA Strandb | Number of Chi sequencesc | Strand ratios | |||
ORF DNA |
Non-ORF DNA | Synonymous/ templated | |||||||
| (kb) | % | Leftward | Rightward | ||||||
| Thermophilic | |||||||||
| M.thermoauto- trophicum | 49.70 | 1.751 | 735 | 42 | Top | 8 | 7 | 3 | 1.64 (23:14) |
| . | Bottom | 16 | 6 | 0 | (P=0.139) | ||||
| . | |||||||||
| A. fulgidus | 48.36 | 2.178 | 627.5 | 28.8 | Top | 7 | 12 | 1 | 1.84 (24:13) |
| . | Bottom | 12 | 6 | 0 | (P=0.071) | ||||
| . | |||||||||
A. aeolicus |
43.47 | 1.551 | 1551 | 100 | Top | 6 | 4 | 1 | 0.69 (9:13) |
| . | Bottom | 5 | 7 | 2 | (P=0.394) | ||||
| . | |||||||||
| P. horikoshii | 41.63 | 1.738 | 735 | 42.3 | Top | 1 | 6 | 3 | 3.17 (19:6) |
| . | Bottom | 13 | 5 | 1 | (P=0.009) | ||||
| . | |||||||||
M. jannaschii |
31.43 | 1.665 | 1665 | 100 | Top | 7 | 18 | 2 | 2.67 (32:12) |
| . | Bottom | 14 | 5 | 2 | (P=0.003) | ||||
| Mesophilic | |||||||||
D.radiodurans |
65.75 | 3.2 | 318.2 | 10 | Top | 10 | 30 | 0 | 2.36 (59:25) |
| . | Bottom | 29 | 15 | 2 | (P=0.0002) | ||||
| . | |||||||||
| T. pallidum | 52.88 | 1.138 | 735 | 64.6 | Top | 3 | 11 | 0 | 4.5 (27:6) |
| . | Bottom | 16 | 3 | 0 | (P=0.0003) | ||||
| . | |||||||||
| E. colie | 50.67 | 4.639 | 735 | 16 | Top | 22 | 52 | 0 | 2.90 (113:39) |
| . | Bottom | 61 | 17 | 1 | (P<0.00001) | ||||
| . | |||||||||
| Synechosystis | 47.14 | 3.573 | 735 | 20.6 | Top | 3 | 18 | 2 | 4.22 (38:9) |
| . | Bottom | 20 | 6 | 0 | (P<0.0001) | ||||
| . | |||||||||
| B. subtilis | 44.36 | 4.512 | 735 | 17.4 | Top | 0 | 2 | 1 | 6.0 (6:1) |
| . | Bottom | 4 | 1 | 0 | (P=0.059) | ||||
| . | |||||||||
| C. trachomatis | 41.29 | 1.043 | 735 | 70.5 | Top | 3 | 5 | 0 | 1.8 (9:5) |
| . | Bottom | 4 | 2 | 0 | (P=0.285) | ||||
| . | |||||||||
| M. pneumoniae | 40.01 | 0.816 | 816.4 | 100 | Top | 2 | 7 | 1 | 3.0 (27:9) |
| . | Bottom | 20 | 7 | 0 | (P=0.003) | ||||
| . | |||||||||
| H. influenzae | 38.30 | 1.831 | 735 | 40.2 | Top | 15 | 54 | 3 | 2.88 (98:34) |
| . | Bottom | 44 | 19 | 6 | (P<0.00001) | ||||
| . | |||||||||
| M. genitalium | 31.70 | 0.58 | 580 | 100 | Top | 1 | 7 | 1 | 3.5 (7:2) |
| . | Bottom | 0 | 1 | 0 | (P=0.096) | ||||
| . | |||||||||
| R. prowazekii | 29.00 | 1.112 | 1112 | 100 | Top | 0 | 4 | 0 | Inf (10:0) |
| . | Bottom | 6 | 0 | 0 | (P=0.002) | ||||
| Legend to Table 1
a In cases where entire genomes were not examined, segments from disparate parts of the genome were usually employed: M. thermoautotrophicum, 374431-619430, 928831-1173830, 1364931-1609930; A. fulgidus, 771-210840, 868001-1078070, 1550851-1758260; P. horikoshii, 244931-489930, 881931-1126930, 1329931-1574930; D. radiodurans (see Methods); T. pallidum, 69931-314930, 433931-678930, 776,931-1021930; E. coli (see Table 3); Synechosystis, 286931-531930, 1494431-1739430, 2834931-3079930; B. subtilis, 155401-400400, 1469931-1714930, 3065931-3310930; C. trachomatis, 3431-248430, 286931-531930, 629931-874930; H. influenzae, (see Table 3).b Chi sequences were located either in the top-strand, or the bottom strand, the former being so designated by the GenBank record.c Chi sequences were scored as associated with either leftward-transcribed ORFS, or rightward-transcribed ORFs if their centres overlapped the ORFs. Otherwise they were scored as being in non-ORF DNA. In the case of H. influenzae some Chi-like sequences were also included.d The ratio of occurrence in the mRNA-synonymous strand of DNA relative to occurrence in the mRNA-template strand. The significance of this ratio for each organism was obtained through c 2 analyses (2� 2 contingency table) with the probability of each Chi being randomly located in either the mRNA-synonymous or mRNA template strand being 0.5.e To the values for 735 kb recorded here, may be added the values for the E. coli segment corresponding to nucleotides 1-200000 from Bell et al. (1998). This provides a synonymous strand/template strand distribution ratio of 3.34 (157:47). |
|||||||||
In all but one instance (A. aeolicus), Chi sequences in mRNA-synonymous strands (the sum of occurrences in bottom strands when transcription is leftward, plus occurrences in top strands when transcription is rightward) occur more frequently than in mRNA template strands (the sum of occurrences in top strands when transcription is leftward, plus occurrences in bottom strands when transcription is rightward). Thus for M. jannaschii there are 14 + 18 = 32 Chi sequences in the mRNA-synonymous strand, and 7 + 5 = 12 Chi sequences in the mRNA-template strand. The ratio of these (32:12) is 2.67. The significance of this asymmetrical distribution (c 2 analyses with a 2� 2 contingency table) is high (P = 0.003).
However, only the two thermophiles of lowest (C+G)% show significant bias (M. jannaschii and P. horikoshii). On the other hand, whatever the (C+G)%, most mesophiles show significant bias (exceptions C. trachomatis and M. genitalium). There are relatively few canonical Chi sequences in B. subtilis (an unrelated sequence has Chi-like activity; Chedin et al., 1998), but the asymmetrical distribution of those that occur is of marginal significance (P = 0.059).
3.3. Chi sequences in GT-rich "islands" in E. coli
Most organisms obey Szybalski's transcription direction rule (Bell and Forsdyke, 1999b). This is manifest as an asymmetry of base compositions between the mRNA-synonymous and template strands of DNA. Thus, the asymmetry in distribution of Chi sequences between the two strands might merely reflect this base-compositional asymmetry. However, Tracy et al. (1997b) proposed that Chi sequences occupy distinct GT-rich islands of the size of an average ORF (about 1 kb).
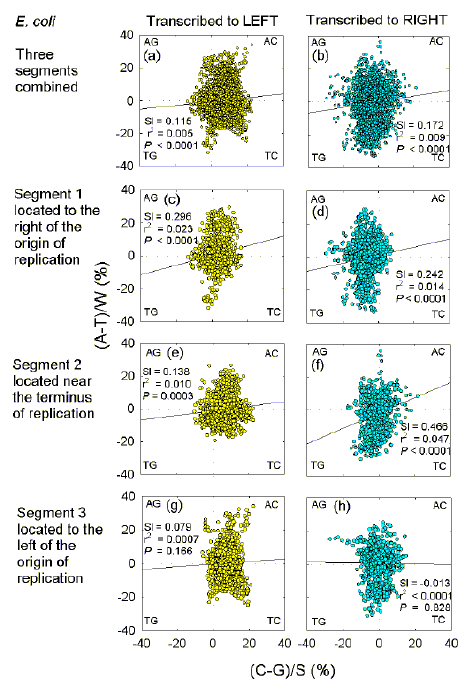
This was examined by seeking differences in base composition, assessed as "Chargaff differences". In "quadrant plots" Chargaff differences for the W bases (A and T) are plotted against Chargaff differences for the S bases (C and G). Following Szybalski's transcription direction rule, one kb sequence windows in the "top" strand whose centres overlap leftward-transcribed ORFs are usually seen to be preferentially enriched in one or more pyrimidines (C, T), whereas one kb sequence windows in the "top" strand whose centres overlap rightward-transcribed ORFs are usually seen to be preferentially enriched in one or more purines (A, G).
Fig. 2 shows quadrant plots for three 245 kb segments from different parts of the circular E. coli genome. This genome has base compositional asymmetries which relate to the origin of replication. The first segment (nt 310941-555940) is from the first part of the genome (located to the right of the origin of replication) where the top-strand tends to be both G-rich and Chi-rich, and rightward-transcribed ORFs predominate. The second segment (nt 1555401-1800400) begins in the region of the terminus of replication, where top-strand G-richness switches to C-richness, leftward-transcribed ORFs become more evident, and Chi sequences begin to predominate in the bottom strand. The third segment (nt 3422021-3667020) is at the other end of this C-rich region, and lies to the left of the origin of replication.
|
|
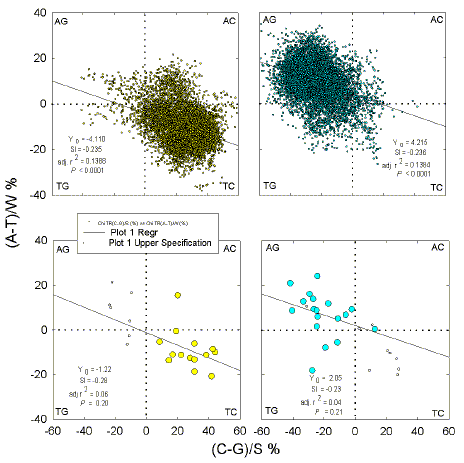
| Fig. 2.
Quadrant analysis of base compositions (expressed as Chargaff differences) in 1 kb windows from the top strand of three segments from the E. coli genome, either combined (a, b), and independent (c-h). The centres of windows overlap either leftward-transcribed ORFs (a, c, e, g), or rightward-transcribed ORFs (b, d, f, h). Each quadrant corresponds to windows enriched for two particular bases (indicated at the corners). Each point corresponds to a window. Listed in each figure are the slopes (Sl) of the least square regression lines (shown as a solid diagonal line), the adjusted squares of the correlation coefficients (r2), and the probabilities (P) that slopes are not significantly different from zero. Only (c) and (f) had significant slopes (P = 0.023 and 0.002, respectively), when the regression analysis used every tenth window to avoid overlapping windows. |
For all three segments combined, a clear preference of leftward ORFs for C and of rightward ORFs for G is apparent (Figs. 2a,b). Although there is a wide scatter of points, linear regression plots have a significant slope, indicating correlations between A-enrichment and C-enrichment, and between G-enrichment and T-enrichment. These correlations are evident in the first and second segments (Figs. 2c-f) but not in the third segment (Figs. 2g,h). Likewise, the G-richness of windows corresponding to rightward-transcribed ORFs is most evident in the first segment (lying to the right of the origin of replication), whereas the C-richness of windows corresponding to leftward-transcribed ORFs is most evident in the third segment (lying to the left of the origin of replication).
|
|
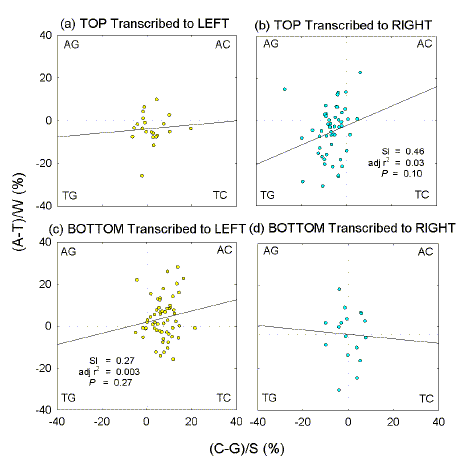
Fig. 3.
Quadrant
analysis of E. coli genome windows with Chi sequences at their centres.
|
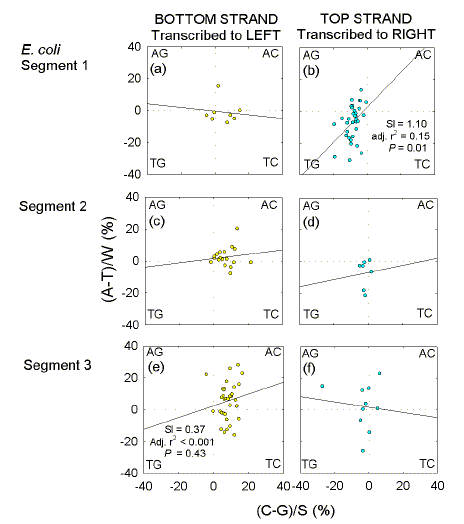
Among these 1 kb windows are some with Chi sequences near their centres ("Chi windows"). Fig. 3 shows that the majority of Chi windows in the top-strand (52) correspond with rightward-transcribed ORFs and tend to be GT-rich, whereas the majority of Chi windows in the bottom strand (61) correspond mainly with leftward-transcribed ORFs and tend to be AC-rich. The minorities of Chi windows in the top strand corresponding to leftward-transcribed ORFs (22), and of Chi windows in the bottom strand corresponding to rightward-transcribed ORFs (17), show no obvious compositional bias. The tendency for GT-richness when corresponding with rightward-transcribed ORFs is particularly apparent in the first segment (Fig. 4b), whereas the tendency for AC-richness when corresponding with leftward-transcribed ORFs is particularly apparent in the third segment (Fig. 4e).
|
|
| Fig. 4. Quadrant analysis for individual E. coli segments of windows with Chi sequences at their centres. Only windows overlapping bottom-strand, leftward-transcribed ORFs, and top-strand, rightward-transcribed ORFs, are shown. (a, b) segment 1. (c, d) segment 2. (e, f) segment 3. |
That Chi windows corresponding to a particular strand and transcriptional orientation are a distinct subset of the total set of windows in the same strand and with the same transcriptional orientation, is shown in Table 2. For example, for the three combined E. coli segments the 52 top strand Chi windows corresponding to rightward transcription are enriched in T (T>A; Chargaff difference value = -5.18se 1.64, which is significantly different from zero; P <0.01). The corresponding total set of 3186 windows shows much less T enrichment (Chargaff difference = -0.74se 0.19, which is also significantly different from zero; P < 0.001). The two sets are significantly different from each other (P = 0.0028; t-test). Similarly, the 61 bottom strand Chi windows corresponding to leftward transcription are enriched in A (A>T; Chargaff difference = 3.80se 1.25, which is significantly different from zero; P < 0.01). The corresponding total set of 3319 windows do not have a significant A enrichment (Chargaff difference = 0.14 se 0.15). Again, the two sets are significantly different from each other (P = 0.001).
| segment number | Strand location of Chi | Chargaff differences (%) | |||||||||||||||||
| . | Chi-containing windows | . | All windows | ||||||||||||||||
| Transcription orientation | Transcriptional orientation | ||||||||||||||||||
| To Left | Nb | Pc |
. | To Right | Nb |
Pc | To Left | Nb | . | To Right | Nb | ||||||||
| 1+2+3 combined | Top | (A-T)/W | T>A | -3.77 se1.51 | 22 | 0.040 | T>A | -5.18 se1.64 | 52 | 0.0028 | A>T | 0.14 se0.15 | 3319 | T>A | -0.74 se0.19 | 3186 | |||
| (C-G)/S | C>G | 1.72 se1.32 | 22 | 0.001 | G>C | -6.64 se0.82 | 52 | 0.0002 | C>G | 5.64 se0.10 | 3319 | G>C | -3.49 se0.11 | 3186 | |||||
| . | . | . | .
|
.
|
|||||||||||||||
| Bottom | (A-T)/W | A>T | 3.80 se1.25 | 61 | 0.001 | T>A | -3.61 se3.03 | 17 | 0.266 | ||||||||||
| (C-G)/S | C>G | 7.29 se0.66 | 61 | 0.020 | C>G | 0.02 se1.36 | 17 | 0.017 | |||||||||||
| . | . | . | |||||||||||||||||
| 1 | Top | (A-T)/W | T>A | -4.94 se3.21 | 9 | 0.072 | T>A | -6.9 se1.83 | 35 | 0.0004 | A>T | 1.02 se0.34 | 849 | T>A | -0.56 se0.29 | 1343 | |||
| (C-G)/S | C>G | 1.71 se1.47 | 9 | 0.606 | G>C | -8.53 se0.70 | 35 | 0.083 | C>G | 2.60 se0.18 | 849 | G>C | -6.97 se0.14 | 1343 | |||||
| . | . | . | .
|
.
|
|||||||||||||||
| Bottom | (A-T)/W | T>A | -1.15 se2.53 | 8 | 0.536 | T>A | -2.57 se10.1 | 4 | 0.704 | ||||||||||
| (C-G)/S | C>G | 4.64 se2.44 | 8 | 0.268 | G>C | -5.60 se1.48 | 4 | 0.605 | |||||||||||
| . | . | . | |||||||||||||||||
| 2 | Top | (A-T)/W | T>A | -3.87 se1.52 | 10 | 0.155 | T>A | -7.30 se3.36 | 7 | 0.238 | T>A | -0.47 se0.21 | 1234 | T>A | -2.26 se0.37 | 909 | |||
| (C-G)/S | C>G | 1.58 se2.47 | 10 | 0.010 | G>C | -1.77 se0.89 | 7 | 0.802 | C>G | 6.27 se0.16 | 1234 | G>C | -1.27 se0.18 | 909 | |||||
| . | . | . | .
|
.
|
|||||||||||||||
| Bottomd | (A-T)/W | A>T | 2.57 se1.42 | 18 | 0.089 | T>A | -7.22 se4.49 | 7 | 0.246 | ||||||||||
| (C-G)/S | C>G | 6.92 se1.36 | 18 | 0.629 | C>G | 3.56 se1.13 | 7 | 0.017 | |||||||||||
| . | . | . | |||||||||||||||||
| 3 | Top | (A-T)/W | A>T | 0.03 se2.78 | 3 | 0.983 | A>T | 2.34 se4.67 | 10 | 0.559 | A>T | 0.15 se0.27 | 1236 | A>T | 0.50 se0.32 | 934 | |||
| (C-G)/S | C>G | 2.24 se3.97 | 3 | 0.074 | G>C | -3.39 se2.90 | 10 | 0.101 | C>G | 7.10 se0.13 | 1236 | G>C | -0.63 se0.17 | 934 | |||||
| . | . | . | . |
. |
|||||||||||||||
| Bottom | (A-T)/W | A>T | 5.57 se1.92 | 35 | 0.001 | T>A | -0.08 se2.91 | 6 | 0.886 | ||||||||||
| (C-G)/S | C>G | 8.08 se0.71 | 35 | 0.222 | G>C | -0.35 se2.62 | 6 | 0.896 | |||||||||||
| Legend to Table 2 a Chargaff differences (%) are presented together with the standard
error of the mean (se). b N = number of windows.c Probabilities (P) that mean base compositions (assessed as Chargaff differences) of 1 kb windows with Chi sequence at their centres, do not differ significantly from those of the corresponding total set of windows (t-test). For further details please see text.d One Chi sequence omitted from the analysis was in an intergenic region between two leftward-transcribed ORFs. |
|||||||||||||||||||
These trends are also evident in the individual segments. For the first segment, the T-excess corresponding to top-strand rightward-transcribed ORFs is most significant (P = 0.0004). For the second segment, the C-excess corresponding to top-strand leftward-transcribed ORFs is most significant (P = 0.010). For the third segment, the A excess corresponding to bottom-strand leftward-transcribed ORFs is most significant (P = 0.001).
3.4. No Chi sequences in GT-rich "islands" in M. jannischii
Thus Chi sequences in E. coli were observed to occupy a distinct subset of 1 kb windows, usually with GT-rich top-strands in the case of top-strand Chis, and with AC-rich top-strands in the case of bottom-strand Chis. This criterion was used to further characterize Chi or Chi-like sequences in other organisms. Whereas E. coli obeys Szybalski's transcription direction rule mainly with respect to the S bases, thermophilic bacteria obey with respect to both the S bases (C and G) and the W bases (Lao and Forsdyke, 2000). In the case of the thermophile M. jannaschii, AC-rich windows are relatively rare in leftward-transcribed regions (see top right quadrant of Fig. 5a), and GT-rich windows are relatively rare in rightward-transcribed regions (see bottom left quadrant of Fig. 5b). These windows are not selectively occupied by Chi sequences (Figs. 5c, d). The most significant difference (P = 0.13) between Chi windows (corresponding to a particular strand and transcription direction) and the total set of windows (corresponding to the same strand and transcription direction), was a slight decrease in the frequency of A bases in the top strand for rightward transcribed ORFs (Chargaff differences 9.92 se 0.09 for all 8202 rightward windows and 7.01 se 2.41 for the corresponding 18 Chi windows). Thus, in this organism there is no clear evidence that Chi windows form a distinct subset.
|
|
Fig. 5. Quadrant analysis of the entire M. jannaschii
genome.
Regression lines are for points corresponding to leftward-transcribed ORFs (a, c), and to rightward-transcribed ORFs (b, d). Y0 is the value of the intercept on the Y axis. Other details are as in previous figures. In (a) and (b) slopes remained significantly different from zero when every tenth point was employed for the regression analysis to avoid overlapping windows. |
3.5. Chi distribution relative to replication and transcription directions
In initial studies of short E. coli segments, Burland et al. (1993) noted that the distribution of Chi sequences correlated independently with the directions both of replication and transcription. However, in H. influenzae Sourice et al. (1998) found only weak evidence for a correlation with replication direction. From studies with single short segments of the genomes of E. coli (nt 1-200000) and H. influenzae (nt 1-350000), the distribution of Chi or Chi-like sequences appeared to correlate better with the direction of transcription than with direction of replication (Bell et al., 1998). Table 3 shows an extension of the latter work to three segments from each organism, the first and third of which are located on either side of the origin of replication, and the second of which is located in the regions either of the origin of replication (H. influenzae) or of the terminus of replication (E. coli). In view of uncertainty regarding locations of origins and terminations of replication, data for Chi distribution with respect to replication direction are omitted in the case of the second segments. For the first and second segments it is apparent for both organisms that correlation with direction of transcription is at least as good as the correlation with direction of replication. Indeed, for the third E. coli segment which lies to the left of the origin of replication, the ratio of occurrences in mRNA synonymous strands relative to template strands (5.0) exceed the ratio of occurrences in leading strands of replicating DNA relative to lagging strands (3.15).
| Bacterium | segment numbera | segment location | Number of ORFs | DNA strand | Number of Chi sequences | Strand ratios | |||||
| Transcription direction | Total | ORF DNA | Non- ORF DNA |
Transcription | Replicationb | ||||||
| Left | Right | Left | Right | Synonymous/ template | Leading/
lagging |
||||||
| . | |||||||||||
| H. influenzae | 1+2+3 combined | . | 315 | 326 | Top | 72 | 15 | 54 | 3 | 2.88 (98:34) | . |
| . | Bottom | 69 | 44 | 19 | 6 | . | |||||
| . | |||||||||||
| 1 | Left of origin | 117 | 103 | Top | 11 | 2 | 9 | 0 | 2.54 (28:11) | 2.64 (29:11) | |
| [103181- 348180] |
. | Bottom | 29 | 19 | 9 | 1 | . | ||||
| . | |||||||||||
| 2 | Origin of replication | 103 | 102 | Top | 29 | 6 | 21 | 2 | 3.45 (38:11) | ||
| [580931- 825931 |
. | Bottom | 27 | 17 | 5 | 5 | . | ||||
| . | |||||||||||
| 3 | Right of origin | 95 | 121 | Top | 32 | 7 | 24 | 1 | 2.67 (32:12) | 2.46 (32:13) | |
| [1224931- 1469930] |
. | Bottom | 13 | 8 | 5 | 0 | . | ||||
| . | |||||||||||
| E. coli | 1+2+3 combined |
. | 372 | 343 | Top | 74 | 22 | 52 | 0 | 2.90 (113:39) | |
| . | Bottom | 79 | 61 | 17 | 1 | ||||||
| . | |||||||||||
| 1 | Right of origin | 94 | 140 | Top | 44 | 9 | 35 | 0 | 3.31 (43:13) | 3.67 (44:12) | |
| [310941- 555940] |
. | Bottom | 12 | 8 | 4 | 0 | . | ||||
| . | |||||||||||
| 2 | Terminus of Replication | 131 | 106 | Top | 17 | 10 | 7 | 0 | 1.47 (25:17) | ||
| [1555401- 1800400] |
. | Bottom | 26 | 18 | 7 | 1 | . | ||||
| . | |||||||||||
| 3 | Left of origin | 147 | 97 | Top | 13 | 3 | 10 | 0 | 5.00 (45:9) | 3.15 (41:13) | |
| [3422021- 3667020] |
. | Bottom | 41 | 35 | 6 | 0 | . | ||||
| Legend to Table
3 a The sequence limits of each segment are shown in parenthesis.b Uncertainties in precise locations of the origins or terminations of replication, make it difficult to assign Chi sequences in the second segments as corresponding to the leading or lagging strands of replicating DNA. However, assuming the origin of replication to be at nt 603000 in the circular H. influenzae genome, segment 2 includes the origin so that in its left part the bottom strand is identical in sequence to the leading strand, and in the right part the top strand is identical in sequence to the leading strand. On this basis, the leading/lagging strand ratio of the distribution of Chi sequences in segment 2 is 1.43 (33:23). In E. coli there are seven experimentally characterized "Ter" (termination) sites spread over 25% of the genome. The "Dif" site (1588774-1588801) is where recA-independent recombination may occur to resolve the two daughter molecules after replication. |
|||||||||||
It should be noted that Chi distribution among leftward or rightward ORFs is not merely explained by the relative availabilities of these two classes of ORFs (Table 3). For example, in the first segment of H. influenzae there are 117 leftward ORFs and 103 rightward ORFs, yet Chi sequences in the top strand predominate in rightward ORFs (2:9; P = 0.02), and the predominance in leftward ORFs of Chi sequences in the bottom strand is about double that expected from ORF distribution (19:9 as opposed to 117:103; P = 0.12).
| 4. Discussion
4.1. Mechanism of homologous recombination A new view, arising from studies of homologous recombination in eukaryotes, is that recombination follows an initial homology search, which may involve exploratory "kissing" interactions (Eguchi et al., 1991) between the tips of single-strand DNA loops extruded from intact DNA duplexes (Kleckner and Weiner, 1993; Forsdyke 1996). Only when such homology has been "tested" through formation of paranemic joints would there be a commitment to recombination (strand-breakage). The initial homology search might involve special regions of DNA, recognized as "hot-spots" or "islands of preferred DNA pairing" (Tracy et al., 1997b). Consistent with this, single-strand loops are gaining recognition as important intermediates in the initiation phase of recombination in bacteria (Anderson and Kowalczykowski, 1998). Bacterial RecA can connect two intact DNA duplexes, which should facilitate homology recognition (Leger et al., 1998; Zaitsev and Kowalczykowski, 1999), and a human RecA homolog has been shown to form homologous joints in the absence of net strand exchange (Gupta et al., 1999). In homologous recombination, an "island of preferred pairing" might suffice to guide certain enzymes of recombination to a region where subsequent strand breakage would occur. However, enzymes such as RecA are also involved in the repair of double-strand breaks. In this case, the initial break might occur at some distance from an "island", so that enzymes recognizing the break (e.g. the RecBCD complex) would have to translocate along the DNA towards an "island", where RecA and other enzymes would then be locally recruited. The "recombination island" proposal emerged at a time when Chargaff difference analysis was revealing genomes as consisting of distinct gene-sized compositional domains characterized by base compositions often in accord with Szybalski's transcription direction rule. However, in one organism (Drosophila melanogaster) which possesses a genome sectored into regions where genes are compactly arranged prokaryote-style and regions where genes are interrupted by introns mammalian-style, the compositional domains were found in both sectors (Dang et al., 1998). Indeed, introns and intergenic DNA of mammals show compositional domains (patterns of Chargaff differences) similar to those in protein-coding regions. This suggested that these domains might be related to some function other than the genic function, perhaps recombination (Bell and Forsdyke, 1999a,b). 4.2. Evolution of recombination Both the bacterial RecA protein and its eukaryotic homologs prefer GT-rich sequences (Tracy et al., 1997a). Furthermore, GT-rich loci are recombinogenic in various organisms (e.g. Jeffreys et al., 1985). Thus, Tracy et al. (1997b) proposed that Chi sequences initially evolved in GT-rich domains which already possessed a low level of intrinsic hot-spot activity. In support of this, they found sequence regularities extending 400 bases on either side of Chi sequences, suggesting a fundamental GT-rich "recombination island" size of about 1 kb, which is about the size both of the average ORF, and of sequence windows at which deviations from Chargaff's second parity rule are maximum when comparing natural with the corresponding shuffled sequences (Bell and Forsdyke, 1999a). Accordingly, it was proposed that early genomes evolved as a mosaic of regions of distinct base composition to facilitate, through loop-loop kissing interactions the interstrand and intrastand "accounting" necessary for recombination repair (Bell and Forsdyke, 1999b). From studies of "Chargaff differences" it was suggested that Chi was critically orientated with respect to transcription direction (Bell et al., 1998), and that the "recombination islands" of Tracy et al. (1997b) had become "recombination/transcription" islands in regions where transcriptional activity had evolved. The adaptive value of recombination was likely to have become evident in the early "RNA world" prior to the development of protein-synthesis (Bell and Forsdyke, 1999b). Thus, early genomes may have contained recombinational domains of varying base compositions, and the transcriptional function may have later imposed upon this recombinational mosaic, prior to the evolution of protein-encoding potential. While we do not know at what stage Chi functions evolved, it seems unlikely that the small Chi octomer was itself sufficient for early recombinational events. More likely, Chi-encoding potential and protein-encoding potential had to adapt independently to the preexisting compositional bias of recombination domains (as well as to the species-specific (C+G)% bias; Forsdyke, 1996). In this light we should reexamine the work of two groups who recently criticised the "recombinational island" suggestion of Tracy et al. (1997b), on the grounds that the frequency and distribution of Chi can be "accounted for" (Colbert et al., 1998), or "explained" (Biaudet et al., 1998) by a preexisting codon usage bias. The "islands of preferred DNA pairing" are held to be merely "oceans of ORFs" (Colbert et al., 1998). However, on the basis of an initial study of a 136 kb segment of the E. coli genome, Burland et al. (1993) argued against an influence of codon usage. We would further argue that codon usage no more explains Chi frequency and distribution than that the disappearance of storks from the rooftops in Holland in the war years explained the concomitant decrease in the human birth rate. Just as these events were both likely to be due to the disturbance of warfare, so a more fundamental cause, the compositional bias of preexisting recombinational domains is likely to have created a genomic environment to which both Chi-encoding potential and protein-encoding potential had to adapt. Colbert et al. (1998) further argue that "Except for the sequence of Chi and its orientation-dependence, no deterministic features of Chi sites are apparent". However, in addition to strand orientation (top or bottom strand), Chi sequences selectively appear in leftward-transcribed ORFs in the bottom strand and in rightward-transcribed ORFs in the top strand (Burland et al., 1993; Bell et al., 1998). This provides a novel deterministic feature of Chi sites, which is the basis of the present analysis. While we agree that in bacteria "recombination islands are equivalent to ORFs" (Colbert et al., 1998), Chargaff difference analysis using 1 kb window sizes provides evidence, at least for E. coli, that Chi windows may correspond to a distinct subset of ORFs (Table 2). To this extent, the "island" proposal is supported. We also note that four Chi-like sequences (GTTGGTGG) in the H. influenzae segments which we examined, occur in 16S RNA genes, where there is no question of an involvement of codons. Three of these four Chi-like sequences occur in the top strand where, as expected, transcription is to the right, and the other occurs in the bottom strand where, as expected, transcription is to the left. 4.4. Chi sequences in other bacteria In this initial study we have limited ourselves to the canonical Chi sequence, except in the case of H. influenzae Chi-like sequences, which are likely to be functional (Sourice et al., 1998). sequences with Chi-like function, but with little similarity to the canonical Chi sequence, have been identified in some bacteria (Smith, 1997; Chedin et al., 1998). In these cases, perhaps because their ecological niches overlap with that of E. coli, there might be a chance of recombination with E. coli DNA (not-self), so there would have been a selection pressure militating against a similarity of sequences with Chi function (so assisting the preservation of species integrity; Forsdyke 1996). Thus, it is pertinent to ask whether canonical Chi sequences are more frequent in bacteria other than E. coli, particularly if they are likely to occupy different niches. If so, one can examine whether these Chi sequences have deterministic features of Chi other than sequence similarity. Indeed, in many such cases, Chi sequences do locate preferentially to mRNA synonymous strands, as in E. coli (Table 1). The fact that Chi abundance in E. coli appears as a distinct outlier when compared with that of other bacteria (Fig. 1), suggests that the canonical Chi is particular to E. coli. However, when expressed as the observed/expected ratio, Chi frequency in E. coli is of the same order as in most other organisms, and less than that of the AT-rich thermophile M. jannaschii. Some bacteria (A. aeolicus, B. subtilis, T. pallidum) had no more canonical Chi sequences than expected from base composition, and in at least one of these (B. subtilis) the Chi function could be mediated by an independent sequence (Chedin et al. 1998). Since recombination repair may be important for correction of DNA damage, and Chi promotes recombination, it was considered possible that the Chi function might be generally increased in thermophilic and radiation-resistant bacteria. These generally occupy ecological niches different from E .coli, so that recombination with the E. coli would be unlikely. Thus, they might have even more canonical Chi sequences than E. coli. However, when base composition was taken into account, D. radiodurans did not have more canonical Chi sequences than E .coli, although many preferentially located to the mRNA synonymous stands (Fig. 1; Table 1). Furthermore, only two of the five thermophilic bacteria examined showed preferential location of Chi sequences to mRNA-synonymous strands. Thus, in thermophilic bacteria and D. radiodurans, the Chi function may be achieved by sequences which differ from the canonical Chi sequence, or the number of these sequences is sufficient, or the Chi function is unnecessary. |
Acknowledgements
We thank J. Gerlach for assistance with computer configuration, and R. Y. P. Chen, G. A. Hill, E. Y. Joo, and R. G. Rasile for assistance in analysis of unedited sequences of D. radiodurans obtained from the Institute for Genome Research. The Glimmer program was kindly donated by S. Salzberg. The National Research Council of Canada, Academic Press and Elsevier Science gave permission for the inclusion of full-text versions of relevant preceding papers at our internet site (http://www.queensu.ca/academia/forsdyke/bioinfor.htm ).
Anderson, D. G., Kowalczykowski, S. C., 1998. SSB protein controls RecBCD enzyme
nuclease activity during unwinding: a new role for looped intermediates. J. Mol. Biol. 282, 275-285.
Bell, S.J., Chow, Y.C., Ho, J.Y.K., Forsdyke, D.R., 1998. Correlation of Chi orientation with transcription indicates a fundamental relationship between recombination and transcription. Gene 216, 285-292.
Bell, S.J., Forsdyke, D.R., 1999a. Accounting Units in DNA. J. Theor. Biol. 197(1), 51-61.
Bell, S.J., Forsdyke, D.R., 1999b. Deviations from Chargaff's second Parity Rule Correlate with Direction of Transcription. J. Theor. Biol. 197, 63-76.
Biaudet, V., El Karoui, M., Gruss, A., 1998. Codon usage can explain GT-rich islands surrounding Chi sites on the Escherichia coli genome. Molec. Microbiol. 29, 661-669.
Burland, V., Plunkett, G., Daniels, D. L., Blattner, F. R., 1993. DNA sequence and analysis of 136 kilobases of the Escherichia coli genome: organizational symmetry around the origin of replication. Genomics 16, 551-561.
Chedin, F., Noirot, P., Biaudet, V., Ehrlich, S. D. 1998. A five-nucleotide sequence protects DNA from exonucleolytic degradation by AddAB, the RecBCD analogue of Bacillus subtilis. Molec. Microbiol. 29, 1369-1377.
Colbert, T., Taylor, A. F., Smith, G. R., 1998. Genomics, Chi sites and codons: 'islands of preferred DNA pairing' are oceans of ORFs. Trends Genet. 14, 485-488.
Dabert, P., Ehrlich, S. D., Gruss, A., 1992. The Chi sequence protects against RecBCD degradation of DNA in vivo. Proc. Natl. Acad. Sci. USA 89, 12073-12077.
Dang, K. D., Dutt, P. B., Forsdyke, D. R., 1998. Chargaff difference analysis of the bithorax complex of Drosophila melanogaster. Biochem. Cell Biol. 76, 129-137.
Dixon, D.A., Kowalczykowski, S.C., 1991. Homologous pairing in vitro stimulated by the recombination hotspot Chi. Cell 66, 361-371.
Eguchi, Y., Itoh, T., Tomizawa, J., 1991. Antisense RNA. Annu. Rev. Biochem. 60, 631-652.
Forsdyke, D. R., 1996. Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths". J. Theor. Biol. 178, 405-417.
Gupta, R. C., Folta-Stogniew, E., Radding, C. M., 1999. Human Rad51 can form homologous joints in the absence of net strand exchange. J. Biol. Chem. 274, 1248-1256.
Jeffreys, A. J., Wilson, V., Thein, S. L., 1985. Hypervariable 'minisatellite' regions in human DNA. Nature 314, 67-73.
Kleckner, N., Weiner, B. M., 1993. Potential advantages of unstable interactions for pairing of chromosomes in meiotic, somatic and premeiotic cells. Cold Spring Harbor Sym. Quant. Biol. 58, 553-565.
Kuzminov, A., Schabtach, E., Stahl, F. W., 1994. Chi sites in combination with RecA protein increase the survival of linear DNA in Escherichia coli by inactivating exoV activity of RecBCD nuclease. EMBO. J. 13, 2764-2776.
Leger, J. F., Robert, J., Bourdieu, L., Chatenay, D., Marko, J. F. 1998. RecA binding to a single double-stranded DNA molecule: a possible role of DNA conformational fluctuations. Proc. Natl. Acad. Sci. USA 95, 12295-12299.
Ponticelli, A. S., Smith, G. R., 1992. Chromosomal context dependence of a eukaryotic recombinational hotspot. Proc. Natl. Acad. Sci. USA 89, 227-231.
Rayssiguier, C., Thaler, D., Radman, M. 1989. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 342, 396-401.
Smith, G. R., 1997. Chi sites and their consequences. In: Bruijn, F.J. de, Lipski, J.R., Weinstock, G. M. (Eds.), Bacterial Genomics. Thomson Publishing, New York, pp. 49-66.
Sourice, S., Biaudet, V., El Karoui, M., Ehrlich, S. D., Gruss, A., 1998. Identification of the Chi site of Haemophilus influenzae as several sequences related to the Escherichia coli Chi site. Mol. Microbiol. 27, 1021-1029.
Tracy, R. B., Baumohl, J. K., Kowalczykowski, S. C., 1997a. The preference for GT-rich DNA by the yeast Rad51 protein defines a set of universal pairing sequences. Genes Devel. 11, 3423-3431.
Tracy, R. B., Chedin, F., Kowalczykowski, S. C., 1997b. The recombination hot spot Chi is embedded within islands of preferred DNA pairing sequences in the E. coli genome. Cell 90, 205-205.
Zaitsev, E. N., Kowalczykowski, S. C., 1999. The simultaneous binding of two double-stranded DNA molecules by Escherichia coli RecA protein. J. Mol. Biol. 287, 21-31.
![]()
Return to: Bioinformatics Index (Click Here)
Return to: Homepage (Click Here)
![]()
Posted here in 2000 and last edited 09 Nov 2020 by Donald Forsdyke