| Chargaff’s legacy
Reviewed by Donald R. Forsdyke & James R. Mortimer |
 |
| Chargaff’s legacy
Reviewed by Donald R. Forsdyke & James R. Mortimer |
|
Presented at the Workshop: Neutralism and Selectionism. The End of a Debate. Ischia May 3rd-6th 2000
Gene (2000) 261, 127-137
With copyright permission from Elsevier Science, which allows single copies to be printed for personal use.
![]()
1. Certain structural principles
4. Adaptive value of purine loading
8. The "holy grail" of Romanes and Bateson
![]()
Abstract
Of Chargaff's four rules on DNA base composition, only his first parity rule was incorporated into mainstream biology as the DNA double helix. Now, the cluster rule, the second parity rule, and the GC rule, reveal the multiple levels of information in our genomes and potential conflicts between them. In these terms we can understand
Keywords: Base composition; Chargaff’s rules; Double-stranded RNA; Self/Not-self discrimination; Speciation; Szybalski’s transcription direction rule; Thermophiles |
1. Certain structural principles
Fifty years ago Erwin Chargaff and his colleagues noted "regularities" in the base composition of nucleic acids, which they considered "reflected the existence in all DNA preparations of certain structural principles" (Chargaff, 1950, 1951). In particular, for duplex DNA they identified a species-invariant component of the base composition, — %A = %T and %C = %G. Shortly thereafter, this "first parity rule" was dramatically confirmed by the Watson-Crick double-helix model (Watson and Crick, 1953). However, the Chargaff laboratory made three other fundamental observations on the base composition of DNA, which are only now being incorporated into mainstream biology. Two of these were species-invariant, but one varied with the species. The first species-invariant observation was that individual bases are clustered to a greater extent than expected on a random basis (the "cluster rule"; Chargaff, 1963). Thus: |
"Another consequence of our studies on deoxyribonucleic acids of animal and plant origin is the conclusion that at least 60% of the pyrimidines occur as oligonucleotide tracts [runs] containing three or more pyrimidines in a row; and a corresponding statement must, owing to the equality relationship [between the two strands], apply also to the purines." |
The second species-invariant observation was that Chargaff's first parity rule also applies, to a close approximation, to single-stranded DNA (his "second parity rule"). If the individual strands of a DNA duplex are isolated and their base compositions determined, then %A = %T, and %C = %G (Rudner et al., 1968). Thus it was noted that there is an: |
"equality — even in the separated DNA strands — of 6-amino [A + C] and 6-keto [G + T] nucleotides, in the absence of all other pairing regularities". |
The validity of the rule became clearer when full genome sequences became available. For example, the "top" strand of Vaccinia virus has 63921 As, 63776 Ts, 32010 Cs, and 32030 Gs. The rule holds, albeit less precisely, even when sequences are divided into segments of a few hundred bases (Bell and Forsdyke, 1999a). Finally, there was the observation that the ratio of C + G to the total bases (A+C+G+T) tends to be constant in a particular species, but varies between species (the "GC rule"; Chargaff, 1951, 1979). |
| "DNA is in its composition characteristic of the species from which it is derived. This can … be demonstrated by determining the ratios in which the individual purines and pyrimidines occur …. There appear to exist two main groups of DNA, namely the ‘AT type,’ in which adenine and thymine predominate, and the ‘GC type,’ in which guanine and cytosine are the major constituents." |
The cluster rule was extended by work from Waclaw Szybalski's laboratory showing that base clustering in microorganisms often relates to transcription direction. The "top" strand of part of a DNA duplex which is transcribed contains pyrimidine clusters if transcription is to the left of the promoter, and purine clusters if transcription is to the right of the promoter (Szybalski et al., 1966). For the circular lambdaphage genome, since genes to the left of the origin of replication are transcribed to the left, and genes to the right of this origin are transcribed to the right, the distribution of clusters also relates to the origin of replication (Szybalski et al., 1969). The observation of base clustering did not necessarily imply a local conflict with Chargaff's second parity rule. For example, a run of T residues, might be accompanied by a corresponding number of dispersed A residues, so that %A = %T. However, Oliver Smithies and his colleagues (1981) showed that there are distinct local deviations from the second parity rule, which again correlate with transcription and replication directions. In the circular genome of SV40 virus it was noted that to the left of the origin of replication genes are transcribed to the left, and here %C > % G, whereas to the right of the origin of replication genes are transcribed to the right, and here %G > %C. Thus, clustering can result in local deviations from the second parity rule in favour of the clustered base. When transcription is to the left, the top strand is the mRNA template strand (pyrimidine-rich), and the bottom strand is the mRNA synonymous strand (purine-rich). When transcription is to the right, the top strand is the mRNA synonymous strand (purine-rich), and the bottom strand is the mRNA template strand (pyrimidine-rich). It follows that, whether arising from a gene transcribed to the left or to the right, mRNAs tend to be "purine-loaded". This may be expressed as the "purine-loading index" (Lao and Forsdyke, 2000), which may be calculated from codon-usage tables (Nakamura et al., 1999; Fig. 1). Some intriguing exceptions (i.e. pyrimidine-loading) are discussed elsewhere (Cristillo et al., 1998; Bell and Forsdyke, 1999b; Forsdyke, 2001). |
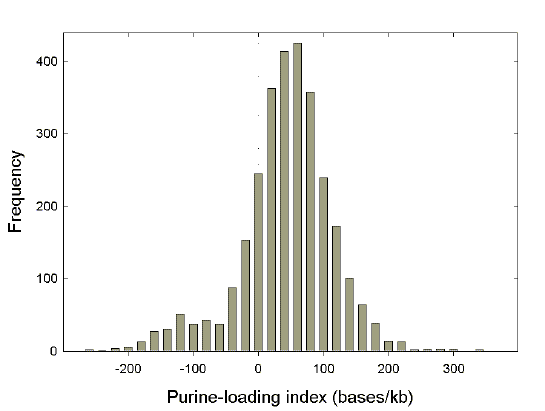 |
Fig. 1. Distribution of purine-loading among species. Purine-loading of coding regions was calculated from codon usage tables for all species represented in the August 1999 release of the GenBank database by more than three genes and more than 2500 bases. The purine-loading index (bases/kb) for a particular species was calculated as the sum of 1000[(G-C)/N] and 1000[(A-T)/N], where G, C, A, and T correspond to the numbers of individual bases, and N corresponds to the total number of bases, in the codon usage table. This measure of the purine-loading of RNAs disregards 5’ and 3’ non-coding sequences, including poly(A) tails. The value for all human genes (excluding mitochondria) is 42 bases/kb, meaning that, on average, there are 42 more purines than pyrimidines for every kilobase of coding sequence. The shoulder with negative purine-loading values (i.e. pyrimidine-loading) corresponds mainly to mitochondrial genes. |
Smithies used non-overlapping "windows" of approx. 0.1 kb to examine base composition. Bases were counted in a window and then the window was moved along the sequence, and the count repeated. Thus a base-composition profile for a genomic region was constructed. When sequences of much larger genomes became available in the 1990s, Donald Forsdyke and colleagues showed that a window of 1 kb can identify more precisely the locations of genes (open reading frames) and their transcriptional orientation (Dang et al., 1998; Bell and Forsdyke, 1999a, b; Fig. 2). Furthermore, Jean Lobry showed that the relationship to the origin of replication is a feature of several microbial genomes ("skew analysis"; Frank and Lobry, 1999; Rocha et al., 1999). |
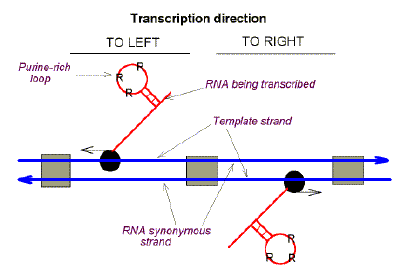 |
Fig. 2. Szybalski's transcription direction rule, evaluated as "Chargaff differences" (deviations from Chargaff's second parity rule). Heavy blue horizontal arrows refer to the "top" and "bottom" strands of duplex DNA. Grey boxes refer to intergenic DNA. Black balls represent RNA polymerases with thin horizontal arrows indicating the direction of transcription. In the case of leftward transcription the Chargaff difference for the top strand is in favour of pyrimidines (Y). In the case of rightward transcription the Chargaff difference for the top strand is in favour of purines (R). It follows that RNAs tend to be purine-loaded. When applied to uncharted DNA, Chargaff difference patterns provide clues to the location of genes and their transcriptional orientation. |
Chargaff's main interest in the base cluster phenomenon was that, prior to the emergence of nucleic acid sequencing technology, it provided some measure of the uniqueness of the base order of a nucleic acid. Szybalski’s main interest was the possibility that the clustering played a role in the control of transcription. This implied an evolutionary selection pressure for clustering so that organisms with clusters would better control transcription than organisms which did not have clusters. However, following a better understanding of Chargaff’s second parity rule as a reflection of nucleic acid secondary structure (see Section 3), it was recognized that in many cases a selection pressure for clustering was likely to have arisen at the post-transcriptional level. |
Chargaff’s first parity rule for duplex DNA was consistent with a base on one strand of the Watson-Crick duplex requiring a complementary base on the other strand of the duplex. By extrapolation, the existence of a parity rule for single strands of nucleic acid (Chargaff’s second parity rule), suggested intrastrand base pairing. At least by virtue of the composition of the stems in stem-loop secondary structures there should be an approximate equivalence between the Chargaff base pairs. Do genomes have the potential to form such secondary structures? What adaptive forces (if any) could have created them? These questions began to be addressed when the genomic sequences of various bacterial viruses (bacteriophages) first became available in the 1970s. It became evident that genomes contain multiple levels of information, and that some forms of information conflict with others (Grantham, 1972; Grantham et al., 1985, 1986). In some phages the genome is RNA (e.g. R17, MS2), and in others the genome is DNA (e.g. T4). In classical Darwinian terms it was assumed that bacteriophages which encode optimal proteins are best adapted to their environment. Thus, the environment acting on virus proteins would have selected for survival the "fittest" viruses whose genes encoded optimal proteins. However, it was observed that the base sequence seems to serve the needs of nucleic acid structure just as much as the protein-encoding function. Indeed, the needs of nucleic acid structure are sometimes served better than those of the protein-encoding function. Since bacteriophages do not encode rRNAs and tRNAs, the possibility arose that it was the structure of mRNAs which was of selective importance. Winston Salser noted in 1970: |
"RNA phage R17 has very extensive regions of highly ordered base pairing. It has seemed likely that this might be necessary to allow phage packaging. Bernice Ricard and I were therefore somewhat surprised to find that T4 messengers [mRNAs], which do not have to be packaged, also have a very large amount of secondary structure. …Our results suggest that a high degree of secondary structure may be important in the functioning of most mRNA molecules. Because of the very high Tm's [temperature at which the "melting" of secondary structure is half maximum] we do not think that the base pairing seen is random. The possible functions of such extensive regions of base-pairing are unknown." |
In 1972 Andrew Ball (Ball, 1972, 1973a,b) went further noting that: |
"The selection pressure for specific base pairing in a messenger RNA severely limits its coding potential", … so that … "there is a pressure for some amino acid sequences to be selected according to criteria which are distinct from the structure and function of the protein they constitute." |
This conclusion was supported by better algorithms for calculating RNA secondary structure (Jaeger et al., 1990), which showed that for many mRNA sequences the energetics of the folding of the natural sequence are better than those of the corresponding shuffled sequence (i.e. one would have to shuffle and fold many times to arrive by chance at a structure approaching the stability of the natural sequence; Le and Maisel, 1989; Forsdyke, 1995a; Seffens and Digby, 1999). It appeared that "the hand of evolution" had arranged the order of bases to support mRNA structure, sometimes at the expense of the coding function. Although abundantly present in the cytoplasm, tRNAs and rRNAs are encoded by a relatively small part of microbial genomes. In microorganisms the sequence of mRNAs, as an RNA entity, are more representative of the genome. If many mRNAs have highly significant secondary structure, then the corresponding genomic regions should also have this potential. Indeed, the primary evolutionary pressure for the elaboration of mRNA secondary structure might have been at the genomic level rather that at the mRNA level. If so, regions of a genome which are not transcribed into mRNAs might also demonstrate potential for secondary structure. When folding algorithms were applied to the sequences of individual DNA strands, it was found that there is indeed considerable potential for secondary structure. Stem-loop potential in DNA is not restricted to regions encoding mRNA (or rRNA or tRNA), but is also present in introns and in intergenic DNA. Stem-loop potential, greater than that of the corresponding shuffled sequence, is diffusely distributed throughout the genomes of all species examined (Forsdyke, 1995a,b,c; Heximer et al., 1996). The conflict with the protein-encoding function was found to be particularly apparent in the case of genes evolving rapidly under positive Darwinian selection (Forsdyke, 1995b; 1996a). In these cases the pressure to adapt the protein sequence has been so powerful that base order has not been able to support stem-loop potential. The natural protein-encoding sequence has less stem-loop potential than the corresponding shuffled sequence. Stem-loop potential is then diverted to introns, which are more conserved than the surrounding exons. Intron stem-loop potential is greater than in the corresponding shuffled sequence. This supports an explanation for the early origin of introns, which postulates that protein-encoding potential was imposed on prototypic genomes already supporting stem-loop potential (Forsdyke 1995a, b, c). At least by virtue of the stems in DNA stem-loop structures, it follows that there would have been an evolutionary pressure for single-strands of DNA to have approximate equivalences of the Watson-Crick pairing bases. Thus, Chargaff's second parity rule is consistent with single strands of DNA having considerable potential for forming secondary structure. The possible adaptive value of this secondary structure at the genomic level will be discussed after first considering the adaptive value of purine-loading at the post-transcriptional level. |
4. Adaptive value of purine loading
An important implication of what is now called "Szybalski's transcription direction rule" is that RNAs, in
general, tend to be purine-loaded (Figs 1, 2). This observation is suggested by Chargaff's
early work on the base composition of total RNA from various species, but his data would
then have mainly reflected the compositions of the most abundant RNA form, the ribosomal
RNAs (rRNAs; Chargaff, 1951; Elson & Chargaff, 1955). When purine-loading was found to
apply to RNAs in general, the possibility arose that the selection pressure for this might
have arisen, not at the transcription level, but post-transcriptionally at the level of
individual RNA molecules. Given that mRNAs tend
where in the structures would purine clusters be found? Since for base-pairing purine clusters must be matched with complementary pyrimidine clusters, and since pyrimidine clusters are scarce in mRNAs, purine clusters should occupy the unpaired regions of mRNA secondary structures, primarily the loops. Indeed, this is where they are found in calculated structures (Bell and Forsdyke, 1999b). Why should it be advantageous for an mRNA to "load" its loops with purines? A possible answer to
this question derived from the studies of Jun-ichi Tomizawa and his colleagues on the way
"sense" and "antisense"
RNA molecules interact, prior to forming a double-strand duplex molecule (dsRNA; Eguchi et
al., 1991; Bull et al., 1998). It was found that sense sequences in RNA search out
complementary antisense sequences through "kissing"
interactions between the tips of the loops of stem-loop structures. If Watson-Crick
base-pairing between the loops is achieved, the interaction may then proceed to the
formation of dsRNA. Thus, if RNAs were generally purine-loaded, "kissing" interactions would decrease, and hence the
probability of forming dsRNA would decrease. How could failure
to form dsRNA be of selective advantage? This will be considered in two steps.
The "distraction hypothesis" (Bell & Forsdyke, 1999b) points out that the physico-chemical state of the "crowded" cytosol (Fulton, 1982; Forsdyke, 1995d) is likely to be highly conducive to a most fundamental interaction, that between the codons of mRNAs and anticodons at the tips of loops in tRNAs. This results in transient formation of dsRNA segments, which normally would not exceed five base pairs (Bossi and Roth, 1980). However, a cytosolic environment which favours mRNA-tRNA kissing interactions would also favour mRNA-mRNA kissing interactions. This "distraction" could have impeded mRNA translation and hence could have decreased the rate of protein synthesis. If, when not seriously conflicting with their coding role, mRNAs had accepted purine mutations in loop regions, then this "purine-loading" would have diminished mRNA-mRNA interactions and increased the efficiency of protein synthesis. In some cases, so important has been the need to accept purine mutations, even the coding role appears to have been compromised (Rocha et al., 1999; Lao and Forsdyke, 2000). When this compromise is not possible, then the length of the protein can be increased by inserting simple sequence repeats of amino acids with purine-rich codons between functional domains (Cristillo et al., 1998; Forsdyke, 2001). The presence of mechanisms to avoid inadvertant formation of extensive segments of dsRNAs as a result of interactions between self-RNAs, leaves open the possibility of the evolution of mechanisms utilizing dsRNA as an intracellular alarm against not-self-RNAs. This was first recognized in the interferon response by which a cell infected by a virus communicates that fact to other cells of an organism. It was found that dsRNA was a most powerful inducer of interferon and of other antiviral adaptations (Isaacs, 1962; Ehrenfeld and Hunt, 1971; Marcus, 1983). However, Phillip Sharp (1999) noted that "it remains a mystery how cells treated with interferon specifically suppress the translation of viral mRNAs in their cytoplasm and not cellular mRNAs". Thus, as with so many problems in biology, the self/not-self discrimination aspect appears central. Recently, dsRNA has been found to mediate intracellular alarms in a variety of plants and animals infected with intracellular pathogens; these pathogens, in turn, have evolved mechanisms to inactivate components of the alarm system (Voinnet et al., 1999; Fire, 1999). Although the mechanism of dsRNA formation is still uncertain (Hamilton and Baulcombe, 1999), one explanation for how an exogenous agent is recognised as not-self and triggers the formation of dsRNA challenges our usual way of regarding the mRNA population of a cell. This population seems obviously heterogeneous because it consists of multiple mRNA species each encoding a different product, — a powerful viewpoint which preempts the search for alternative explanations. However, if we consider the possibility that heterogeneity can serve more than one purpose, then we can regard the heterogenous intracellular mRNA population in the same way as we regard the heterogenous extracellular immunoglobulin population. The latter constitutes the first barrier against foreign pathogens ("not-self"), which select from among the immunoglobulins (which have been randomly generated and then pre-selected for non-reaction with self antigens) those with specificity for the coat antigens of the pathogen. In the case of a virus, the coat is relinquished at the time of entry into a cell, and the cell’s first possible line of intracellular defence would be to recognize the foreign nucleic acid as "not-self", either in its genomic form, or in the form of an early transcript. This implicates dsRNA. Perhaps a segment of a particular host mRNA species, because it happened to have sufficient sequence complementarity, would form a double-stranded segment with viral RNA of sufficient length (at least two helical turns) to trigger an alarm response (Izant and Weintraub, 1984; Melton, 1985; Robertson and Mathews, 1996; Tian et al., 2000). Thus, the heterogenous mRNA population can be seen as consisting of "RNA antibodies", which have been preselected over evolutionary time not to react with "self" RNAs, while retaining, and if possible developing, the potential to react with "not-self" RNAs. Since, due to failure of transcription termination, a low level of transcription of extra-genic DNA is possible (Heximer et al., 1998), the maximum potential repertoire of "RNA antibodies" is limited only by genome size. A second line of intracellular defence would be recognition of the translation products of virus genes; a mechanism for this form of self/not-self discrimination is discussed elsewhere (Forsdyke, 1995d). |
We propose above that in some circumstances evolutionary selective pressures have acted to preserve nucleic acid secondary structure, sometimes at the expense of an encoded protein. That this might also apply to the species-dependent component of the base composition, (C+G)%, arose from Naboru Sueoka's demonstration in 1961, before the genetic code was deciphered, that the amino acid composition of the proteins of microorganisms is influenced, not just by the demands of the environment on the proteins, but also by the base composition of the genome encoding those proteins. The observation has since been abundantly confirmed in a wide variety of animal and plant species (Lobry, 1997). Sueoka (1961) further pointed out that for individual "strains" of Tetrahymena the (C+G)% (referred to as "GC") tends to be uniform throughout the genome: |
"If one compares the distribution of DNA molecules of Tetrahymena strains of different mean GC contents, it is clear that the difference in mean values is due to a rather uniform difference of GC content in individual molecules. In other words, assuming that strains of Tetrahymena have a common phylogenetic origin, when the GC content of DNA of a particular strain changes, all the molecules undergo increases or decreases of GC pairs in similar amounts. This result is consistent with the idea that the base composition is rather uniform not only among DNA molecules of an organism, but also with respect to different parts of a given molecule." |
Again, this observation has been abundantly confirmed for a wide variety of species (Muto and Osawa, 1987), although many organisms considered higher on the evolutionary scale have their genomes sectored into regions of low or high (C+G)% (Bernardi and Bernardi, 1986; Bernardi 2000; see Section 9). Sueoka (1961) also noted a link between (C+G)% and reproductive isolation for strains of Tetrahymena: |
"DNA base composition is a reflection of phylogenetic relationship. Furthermore, it is evident that those strains which mate with one another (i.e. strains within the same 'variety') have similar base compositions. Thus strains of variety 1 ..., which are freely intercrossed, have similar mean GC content." |
When the genetic code was deciphered in the early 1960s, it was observed that there are more codons than amino acids, so that most amino acids can correspond to more than one triplet codon. This gives some flexibility to a nucleic acid sequence. Sometimes an amino acid can be encoded from among as many as six possible synonymous codons. Walter Fitch (1974) noted that "the degeneracy of the genetic code provides an enormous plasticity to achieve secondary structure without sacrificing specificity of the message". Yet, as outlined above, sometimes even this "plasticity" is insufficient, so that, with the exception of genes under positive Darwinian selection (Forsdyke, 1995b, 1996a), genomic secondary structure ("fold pressure") and (C+G)% "call the tune". Non-synonymous codon changes modify the amino acid sequence, sometimes at the expense of protein structure and function. A protein has to adapt to the demands of the environment, but it also has to adapt to genomic forces which we will show have derived, not from the conventional environment acting upon the convention ("classical") phenotype, but from what we call the "reproductive environment" acting on the "genome phenotype'', or "reprotype". Thus Bernardi and Bernardi noted in 1986 that: |
"The organismal phenotype comprises two components, the classical phenotype, corresponding to the 'gene products', and a 'genome phenotype' which is defined by [base] compositional constraints." |
The issue of which codon was employed in a particular circumstance was considered by Richard Grantham, who noted in 1972 that codon choice was not random in microorganisms, "suggesting a mechanism against [base] composition drift". Observing that "little latitude appears left for ‘neutral’ or synonymous mutations in coliphage codons", he was led to his "genome hypothesis", which specified that undefined adaptive genomic pressure(s) caused changes in base composition and hence in codon choice (Grantham et al., 1986): |
There was also a sense that the coding strategy was of relevance to the most fundamental aspects of an organism's biology: |
"What is the fundamental explanation for interspecific variation in coding strategy? Are we faced with a situation of continuous variation within and between species, thus embracing a Darwinian perspective of gradual separation of populations to form new species …? This is the heart of the problem of molecular evolution." |
Grantham and his colleagues further pointed to the need to determine "how much independence exists between the two levels of evolution" (that of the genome phenotype and of the classical phenotypic) and considered "it is too easy just to say most mutations are neutral". However, non-adaptive "neutralist" explanations gained much support (Filipski, 1990; Sueoka, 1995). Paul Sharp and his colleagues concluded in 1993 that the main factors influencing codon choice are mutational biases and the need for highly expressed genes to be efficiently translated. Although phenotypic adaptive factors such as the need to translate an abundant mRNA efficiently can influence codon choice, genomic factors, identified here as stem-loop potential ("fold pressure") and (C+G)%, play an important and often dominant role. |
The secondary structure of nucleic acids with a high (C+G)% is more stable than that of nucleic acids with a low (C+G)%. GC bonds are associated with a more stable nucleic acid structure than AT or AU bonds. This is reflected in the base composition of RNAs whose structure is vital for their function, namely rRNAs and tRNAs. Free of coding constraints, yet required to form part of the precise structure of ribosomes, rRNAs might more readily accept mutations which increase GC content than do mRNAs. Indeed, the GC content of rRNAs is directly proportional to the normal growth temperature, so that rRNAs of thermophilic bacteria are highly enriched in G and C (Dalgaard and Garret, 1993; Forterre and Elie, 1993; Galtier and Lobry, 1997). However, although optimum growth temperature correlates positively with the GC content of rRNA, it does not correlate similarly with the GC content of genomic DNA, and hence with that of the mRNA populations transcribed from that DNA. The finding of no consistent trend towards a high genomic GC in thermophilic organisms has been interpreted as supporting the neutralist argument that variations in genomic GC are the consequences of mutational biases and are, in themselves, of no adaptive value (Filipski 1990; Galtier and Lobry 1997). However, the finding is also consistent with the argument that genomic GC is too important merely to follow the dictates of temperature, since its primary role is related to other more fundamental adaptations (Bernardi and Bernardi, 1986). Galtier and Lobry (1997) have argued that "any secondary structure that must endure high temperatures requires a high G + C content". This would include both the classical Watson-Crick secondary structure involving inter-strand base pairing, and any secondary structure involving intra-strand base pairing (Murchie et al., 1992). However, the stability of genomic DNA at high temperatures might be achieved in ways other than by an increase in GC content (Bernardi, 2000). These include association with polyamines (Oshima et al., 1990), and relaxation of supercoiling (Friedman et al., 1995). There is no reason to believe that in thermophiles DNA is not able to maintain both its classical duplex structure with H-bonding between opposite strands, and any secondary structures involving intrastrand H-bonding. As we propose (see Section 9), the latter structures would be critical only under certain clearly defined, but selectively very important, circumstances, namely when recombination repair is required. The most enduring DNA secondary structure, even at high temperatures, would be the classical duplex form. |
8. The "holy grail" of Romanes and Bateson
With hindsight it seems that, in identifying (C+G)% as the species variant component of the base composition, Chargaff had uncovered what we might now recognize as the "holy grail" of speciation first postulated in 1886 by Charles Darwin's research associate, George Romanes (Forsdyke, 1999a, b). Romanes had pointed to what we would now call non-genic variations in the germ-line, which would tend to isolate an individual reproductively from other members of its species, but not from members that had undergone the same variation. William Bateson further postulated a non-genic inherited variation, which would remain constant for a species, whereas genic variations could occur within a species. The non-genic variations, in whatever was responsible for carrying hereditary information from generation to generation (not known at that time), would have the potential to lead to species differentiation, so that variant members of a species ("not-self") would not successfully reproduce with members of the main species ("self"). The latter would constitute the "reproductive environment" moulding the genome phenotype (reprotype). Once reproductive isolation was achieved, the natural selection postulated by Darwin would be able to further increase species differentiation by allowing the survival of organisms with advantageous genic variations, and disallowing the survival of organisms with disadvantageous genic variations. These genic variations would affect the classical phenotype. Romanes referred to his holy grail (speciating factor) as an "intrinsic peculiarity" of the reproductive system. Bateson described his holy grail as a speciating factor uniformly attached to the same "residue" as the genes, but distinct from the genes. These are just the properties we find in the (C+G)% (Forsdyke, 1996b, 1998). A metaphor for the role (C+G)% might play in keeping individuals reproductively isolated from each other is provided by the word "dialect" (Grantham et al., 1986). A common language brings people together, and in this way is conducive to sexual reproduction. But languages can vary, first into dialects and then into independent sub-languages. Linguistic differences keep people apart, and this difference in the reproductive environment militates against sexual reproduction. At the molecular level, we see similar forces acting at the level of meiosis. Here paternal and maternal chromosomal homologs align. Forsdyke has proposed that if there is sufficient sequence identity, so that the DNA "dialects" [(C+G)%] match, meiosis is likely to progress through various check-points (Page and Orr-Weaver, 1986), and gametes will be formed. If there is insufficient identity (i.e. the DNA "dialects" do not match) meiosis will fail, gametes will not form, and the individual will be sterile, — a "mule". Thus, the original parental lines will be reproductively isolated from each other, and as such would be defined as distinct species. Changes in the (C+G)% dialects have the potential to initiate speciation (Forsdyke, 1996b). |
In 1922 Muller suggested that the pairing of genes as parts of chromosomes undergoing meiotic synapsis, might provide clues to gene structure and replication: |
"It is evident that the very same forces which cause the genes to grow should also cause like genes to attract each other, ... If the two phenomena are thus dependent on a common principle in the make-up of the gene, progress made in the study of one of them should help in the solution of the other." |
In 1954 he set his students an essay "How does the Watson-Crick model account for synapsis?" (Carlson, 1981). Crick took up the challenge in 1971 with his "unpairing postulate" by which the two strands of the classical DNA duplex would unpair to allow a homology search. Recent work on the meiotic alignment of chromosomes suggests that initiation of speciation involves that aspect of the genome phenotype (reprotype), which is constituted by the genome-wide potential to extrude stem-loops. Those of the maternal and paternal chromosomal homologs may mutually explore each other and test for "self" DNA complementarity, using the "kissing" mechanism of Tomizawa (Eguchi et al., 1991; Hawley and Arbel, 1993; Kleckner, 1997). If sufficient complementarity is found (i.e. the genomes are reprotypically compatible), then crossing over and recombination can occur. The main adaptive value of this would be to provide for the correction of errors in the individual homologs (Winge, 1917; Bernstein and Bernstein, 1991). Where does the (C+G)% "dialect" come into this? It has been observed that small fluctuations in (C+G)% would have a major effect on the ability of duplex DNA molecules to extrude stem-loops and on the pattern of loops which then occur. A very small difference in (C+G)% (reprotypic difference) would mark a meiotically pairing DNA as "not-self". This would impair the kissing interaction with "self" DNA (Forsdyke 1998; Bull et al., 1998), and so would disrupt meiosis and allow divergence between the two parental lines, thus initiating speciation (Forsdyke, 1996b). Consistent with this, direct tests of incipient speciation in the fruit fly (the phenomenon known as Haldane’s rule) implicate differences in DNA per se, rather than in distinct genes, as initiating reproductive isolation (Naveira and Maside, 1998; Forsdyke, 2000a). Similar considerations may apply to the phenomenon of "heteroduplex resistance" between recombining DNAs of different bacterial species (Majewski and Cohan, 1998). Once a speciation process has begun, prezygotic factors and postzygotic factors other than (C+G)% are likely to replace the original difference in (C+G)% as a barrier to reproduction (i.e. a barrier to recombination). In this circumstance, (C+G)% becomes free to adopt other roles, such as the prevention of intragenomic recombination. This could involve intragenomic differentiation of regions of high and low (C+G)% (isochores). These have the potential to "reproductively isolate" (i.e. recombinationally isolate) different parts of the genome. Thus, the attempted duplication of a globin gene into alpha-globin and beta-globin genes might have failed since sequence similarity would favour recombination between the two genes and any incipient differences would be eliminated. However, the duplication appears to have involved relocation to a different isochore with corresponding changes in (C+G)%, so that the two genes became recombinationally isolated. As a consequence of the differences in (C+G)% the corresponding mRNAs utilize different codons for corresponding amino acids, even though both mRNAs are translated in the same cell using the same ribosomes and tRNA populations. Thus, it is unlikely that the primary pressure to differentiate codons arose at the translational level (Grantham, 1986). Isochores would have arisen as a random fluctuation in base composition in a genomic region such that one product of a gene duplication was able to survive for a sufficient number of generations to allow functional differentiation to occur. The regional base compositional fluctuation would then have "hitch-hiked" through the generations on the successful duplicate. |
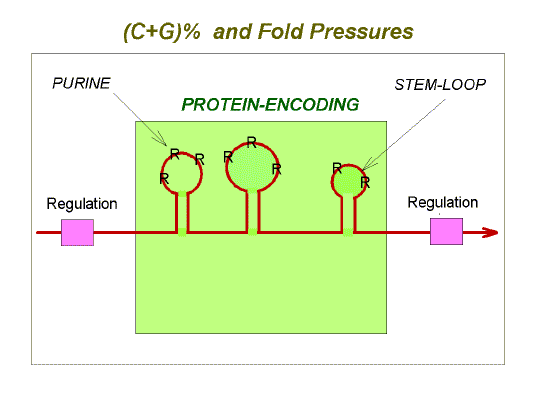 |
Fig. 3. Summary of potentially conflicting evolutionary
pressures as manifest at the level of mRNA (red line with arrow-head).
|
The multiple evolutionary pressures acting on mRNAs and genomes are summarized in Figure 3 and Table 1. It should be noted that changes in genetic fitness (changes in the number of progeny transmitted to future generations), could result from changes in the classical phenotype and/or in the genome phenotype. A mutation which appeared neutral with respect to protein function might nevertheless affect fitness by changing the genome phenotype. It is argued elsewhere that the apparently neutral polymorphism of some intracellular proteins is an adaptation to facilitate intracellular self/not-self discrimination (Forsdyke, 2000b). |
| Table 1 Postulated evolutionary processes leading to multiple levels of information in genomes | ||||
| Environmental selective factors | Selection for mutations which: | Primary effect on DNA function | Biological result | Observed features of modern DNA |
| Classical phenotypic selective factors | Change encoded proteins | None.
|
Change in classical phenotype | Usually change in a base pair in accordance with Chargaff’s first parity rule |
| Competitors with more efficient translation. | Purine-load RNAs | None.
|
Efficient translation with no "self" dsRNA formation | Chargaff's cluster rule and Szybalski's transcription direction rule |
| Mutagens | Promote DNA stem-loop potential | Within species meiotic recombination promoted | Change in genome phenotype for DNA repair | Chargaff's second parity rule |
| Recombinationally "not-self" sexual partners | Impair homology search between DNAs of species members whose sequences are diverging | Meiotic recombination is impaired | Change in genome phenotype for speciation | Chargaff's GC rule |
Chargaff was well aware of the evolutionary implications of his work, writing of "the survival of the fittest nucleic acids" (Chargaff, 1951). However, Biology satisfied with what was called "the modern synthesis" (Carlson, 1981), had not kept pace with Chemistry, and it was difficult for him to explore the implications of his discovery (Chargaff, 1979). Romanes and Bateson, who had challenged Darwinian dogma, had been dismissed by generations of biologists bewitched by genes and all that they promised. To put it mildly, the level of discourse was not constructive.
|
"For historians there remains the baffling enigma of how such distinguished biologists as …W. Bateson … could rest satisfied with such a crassly inadequate theory. ... The irony with which we must now read W. Bateson's dismissal of Darwin is almost painful." |
Despite the overwhelming rhetoric to the contrary, we are now beginning to appreciate the deep truths which Romanes and Bateson were attempting to convey (Forsdyke, 1999a,b; 2000a). When Bateson died in 1926, Chargaff was twenty. He is still with us, – a citizen of the twenty first century. We wish him well. |
Acknowledgements
We thank Richard Grantham and Tim Greenland for a very helpful review of the manuscript, and Jim Gerlach for assistance with computer configuration. Academic Press, Cold Spring Harbor Laboratory Press, Elsevier Science and the National Research Council of Canada gave permission to place full-text versions of some of the above cited papers in Forsdyke's web pages.
References
Ball, L. A. 1972. Implications of secondary structure in messenger RNA. J. Theor. Biol. 36, 313-320.
Ball, L. A. 1973a. Secondary structure and coding potential of the coat protein gene of bacteriophage MS2. Nature New Biol. 242, 44-45.
Ball. L. A. 1973b. Mutual influence of the secondary structure and information content of a messenger RNA. J. Theor. Biol. 41, 243-247.
Bell, S. J., Forsdyke, D. R. 1999a. Accounting units in DNA. J. Theor. Biol. 197, 51-61.
Bell, S. J., Forsdyke, D. R. 1999b. Deviations from Chargaff's second parity rule correlate with direction of transcription. J. Theor. Biol. 197, 63-76.
Bernardi G., Bernardi, G. 1986. Compositional constraints and genome evolution. J. Mol. Evol. 24, 1-11.
Bernardi G. 2000. Isochores and the evolutionary genomics of vertebrates. Gene 241, 3-17.
Bernstein, C., Bernstein, H. 1991. Aging, Sex and DNA Repair. Academic Press, San Diego.
Bossi, L., Roth, J. R. 1980. The influence of codon context on genetic code translation. Nature 286, 123-127.
Bull, J. J., Jacobson, A., Badgett, M. R., Molineux, I. J. 1998. Viral escape from antisense RNA. Molec. Microbiol. 28, 835-846.
Carlson, E. A. 1981. Genes, Radiation and Society. The Life and Work of H. J. Muller. Cornell University Press, Ithaca, NY, pp. 390-392.
Chargaff, E. 1950. Chemical specificity of nucleic acids and mechanism of their enzymic degradation. Experientia 6, 201-209.
Chargaff, E. 1951. Structure and function of nucleic acids as cell constituents. Fed. Proc. 10, 654-659.
Chargaff, E. 1963. Essays on Nucleic Acids. Elsevier, Amsterdam.
Chargaff, E. 1979. How genetics got a chemical education. Ann. N. Y. Acad. Sci. 325, 345-360.
Crick, F. 1971. General model for the chromosomes of higher organisms. Nature 234, 25-27.
Cristillo, A. D., Lillicrap, T. P., Forsdyke, D. R. 1998. Purine-loading of EBNA-1 mRNA avoids sense-antisense "collisions". FASEB. J. 12, A1453.
Dalgaard, J.Z., Garrett, A. 1993. Archaeal hyperthermophile genes. In: Kates, M., Kushner, D. J., Matheson, A. T. (Eds.), The Biochemistry of Archaea (Archaebacteria). Elsevier, Amsterdam, pp 535-562.
Dang, K. D., Dutt, P. B., Forsdyke, D. R. 1998. Chargaff differences correlate with transcription direction in the bithorax complex of Drosophila. Biochem. Cell Biol. 76,129-137.
Dawkins, R. 1983. Universal Darwinism. In: Bendall, D. S. (Ed.), Evolution from Molecules to Man. Cambridge University Press, Cambridge, pp. 405-425.
Eguchi, Y., Itoh, T., Tomizawa, J. 1991. Antisense RNA. Annu. Rev. Biochem. 60, 631-652.
Ehrenfeld, E., Hunt, T. 1971. Double-stranded poliovirus RNA inhibits initiation of protein synthesis by reticulocyte lysates. Proc. Natl. Acad. Sci. USA 68, 1075-1078.
Elson, D., Chargaff, E. 1955. Evidence of common regularities in the composition of pentose nucleic acids. Biochem. Biophys. Acta 17, 367-376.
Filipski, J. 1990. Evolution of DNA sequences. Contributions of mutational bias and selection to the origin of chromosomal compartments. Adv. Mut. Res. 2:1-54.
Fire, A. 1999. RNA-triggered gene silencing. Trends Genet. 15, 358-363.
Fitch, W. M. 1974. The large extent of putative secondary nucleic acid structure in random nucleotide sequences of amino acid-derived messenger-RNA. J. Mol. Evol. 3, 279-291.
Forsdyke, D. R. 1995a. A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol. Biol. Evol. 12, 949-958.
Forsdyke, D. R. 1995b. Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. An application of FORS-D analysis. Mol. Biol. Evol. 12, 1157-1165.
Forsdyke, D. R. 1995c. Relative roles of primary sequence and (G+C)% in determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J. Mol. Evol. 41, 573-581.
Forsdyke, D. R. 1995d. Entropy-driven protein self-aggregation as the basis for self/not-self discrimination in the crowded cytosol. J. Biol. Sys. 3, 273-287.
Forsdyke, D. R. 1996a. Stem-loop potential in MHC genes: a new way of evaluating positive Darwinian selection. Immunogenetics 43, 182-189.
Forsdyke, D. R. 1996b. Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths". J. Theor. Biol. 178, 405-417.
Forsdyke, D. R. 1998. An alternative way of thinking about stem-loops in DNA. A case study of the human G0S2 gene. J. Theor. Biol. 192, 489-504.
Forsdyke, D. R. 1999a. Two levels of information in DNA. Relationship of Romanes' "intrinsic" variability of the reproductive system, and Bateson's "residue" to the species-dependent component of the base composition, (C+G)%. J. Theor. Biol. 201, 47-61.
Forsdyke, D. R. 1999b. The origin of species, revisited. Queen’s Quarterly 106, 112-134.
Forsdyke, D. R. 2000a. Haldane’s rule: hybrid sterility affects the heterogametic sex first because sexual differentiation is on the path to species differentiation. J. Theor. Biol. 204, 443-452.
Forsdyke, D. R. 2000b. Double-stranded RNA and/or heat-shock as initiators of chaperone mode switches in diseases associated with protein aggregation. Cell Stress.Chaperones 5, 375-376.
Forsdyke, D. R. 2001. Search for a Victorian. The Origin of Species, Revisited. McGill-Queen’s University Press, Montreal. (in press)
Forterre, P., Elie, C. 1993. Chromosome structure, DNA topoisomerases, and DNA polymerases in archaebacteria (archeae). In: Kates, M., Kushner, D. J., Matheson, A. (Eds.), The Biochemistry of Archaea (Archaebacteria). Elsevier, Amsterdam, pp 325-345.
Frank, A. C., Lobry. J. R. 1999. Asymmetric substitution patterns: a review of possible underlying mutational or selective mechanisms. Gene. 238, 65-77.
Friedman, S. M., Malik, M., Drlica, K., 1995. DNA supercoiling in a thermotolerant mutant of Escherichia coli. Mol. Gen. Genet. 248, 417-422.
Fulton, A. B. 1982. How crowded is the cytoplasm? Cell 30, 345-347.
Galtier, N., Lobry, J. R. 1997. Relationships between genomic G + C content, RNA secondary structures, and optimal growth temperature in prokaryotes. J. Mol. Evol. 44, 632-636.
Grantham, R. 1972. Codon base randomness and composition drift in coliphage. Nature New Biol. 237, 265-266.
Grantham, R., Greenland, T., Louail, S., Mouchiroud, D., Prato, J. L., Gouy, M., Gautier, C. 1985. Molecular evolution of viruses as seen by nucleic acid sequence study. Bull. Inst. Past. 83, 95-148.
Grantham, R., Perrin, P., Mouchiroud, D. 1986. Patterns in codon usage in different kinds of species. Oxford Surv. Evol. Biol. 3, 48-81.
Hamilton, A. J., Baulcombe, D. C. 1999. A species of small antisense RNA in post-transcriptional gene silencing in plants. Science 286, 950-951.
Hawley, R. S., Arbel, T. 1993.Yeast genetics and the fall of the classical view of meiosis. Cell 72, 301-303.
Heximer, S. P., Cristillo, A. D., Russell, L., Forsdyke, D. R. 1996. Sequence analysis and expression in cultured lymphocytes of the human FOSB gene (G0S3). DNA Cell Biol.15, 1025-1038.
Heximer, S. P., Cristillo, A. D., Russell, L., Forsdyke, D. R. 1998. Expression and processing of G0/G1 Switch Gene 24 (G0S24/TIS11/NUP475) RNA in cultured human blood mononuclear cells. DNA Cell Biol. 17, 249-263.
Isaacs, A. 1962. Antiviral action of interferon. Brit. Med. J. 2, 353-355.
Izant, J. G., Weintraub, H. 1984. Inhibition of thymidine kinase gene expression by anti-sense RNA: a molecular approach to genetic analysis. Cell 36, 1007- 1015.
Jaeger, J. A., Turner, D. H., Zuker, M. 1990. Predicting optimal and suboptimal secondary structure for RNA. Meth Enzymol 183, 281-317.
Kleckner, N. 1997. Interactions between and along chromosomes during meiosis. Harvey Lect. 91, 21-45.
Lao, P. J., Forsdyke, D. R. 2000. Thermophilic bacteria strictly obey Szybalski's transcription direction rule and politely purine-load RNAs with both adenine and guanine. Genome Res. 10, 228-236.
Le, S-Y., Maizel, J. V. 1989. A method for assessing the statistical significance of RNA folding. J. Theor. Biol. 138, 495-510.
Lobry, J. R. 1997. Influence of genomic G + C content on average amino-acid composition of proteins from 59 bacterial species. Gene 205, 309-316.
Majewski, J., Cohan, F. M. 1998. The effect of mismatch repair and heteroduplex formation on sexual isolation in Bacillus. Genetics 148, 13-18.
Marcus, P. 1983. Interferon induction by viruses: one molecule of dsRNA as the threshold for induction. Interferon 5, 115-180.
Mayr, E. 1980. Some thoughts on the history of the evolutionary synthesis. In: Mayr, E.,Provine, W. B. (Eds.), The Evolutionary Synthesis: Perspectives on the Unification of Biology. Harvard Univ. Press, Cambridge, MA, pp. 1-48.
Melton, D. A. 1985. Injected antisense RNAs specifically block messenger RNA translation in vivo. Proc. Natl. Acad. Sci. USA 82, 144-148.
Muller, H. J. 1922. Variation due to change in the individual gene. Am. Nat. 56, 32-50.
Murchie, A. I. H., Bowater, R., Aboul-Ela, F., Lilley, D. M. J. 1992. Helix opening transitions in supercoiled DNA. Biochem. Biophys. Acta 1131, 1-15.
Muto, A., Osawa, S. 1987. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc. Natl. Acad. Sci. USA 84, 166-169.
Nakamura, Y., Gojobori, T., Ikemura, T. 1999. Codon usage tabulated from the international DNA sequence databases: its status 1999. Nucleic Acids Res. 27, 292.
Naveira, H. F., Maside, X. R. 1998. The genetics of hybrid male sterility in Drosophila. In: Howard, D. J., Berlocher, S. H. (Eds.), Endless Forms: Species and Speciation. Oxford Univ. Press, New York, pp. 330-338.
Oshima, T., Hamasaki, N., Uzawa, T., Friedman, S. M. 1990. Biochemical functions of unusual polyamines found in the cells of extreme thermophiles. In: Goldembeg, S. H., Algranati, I. D. (Eds.), The Biology and Chemistry of Polyamines. Oxford University Press, New York, pp 1-10.
Page, A. W., Orr-Weaver, T. L. 1996. Stopping and starting the meiotic cycle. Curr. Opin. Genet. Devel. 7, 23-31.
Robertson, H. D., Mathews, M. B. 1996. The regulation of the protein kinase PKR by RNA. Biochimie 78, 909-914.
Rocha, E. P. C., Danchin, A., Viari, A. 1999. Universal replication biases in bacteria. Mol. Microbiol. 32, 11-16.
Rudner, R., Karkas, J. D., Chargaff, E. 1968. Separation of B. subtilis DNA into complementary strands. III. Proc. Natl. Acad. Sci. USA 60, 921-922.
Salser, W. 1970. Discussion. Cold Spring Harb. Symp. Quant Biol. 35, 19.
Seffens, W., Digby, D. 1999. mRNAs have greater negative folding free energies than shuffled or codon choice randomized sequences. Nucleic Acids Res. 27, 1578-1584.
Sharp, P. A. 1999. RNAi and double-stranded RNA. Genes Devel. 13, 139-141.
Sharp, P. M., Stenico, M., Peden, J. F., Lloyd, A. T. 1993. Codon usage: mutational bias, translation selection, or both? Biochem. Soc. Trans. 21, 835-841.
Smithies, O., Engels, W. R., Devereux, J. R., Slightom, J. L., Shen, S. 1981. Base substitutions, length differences and DNA strand asymmetries in the human Gl and Al fetal globin gene region. Cell 26, 345-353.
Stebbins, G. L. 1980. Botany and the synthetic theory of evolution. In: Mayr, E., Provine, W. B. (Eds.), The Evolutionary Synthesis: Perspectives on the Unification of Biology. Harvard Univ. Press, Cambridge, MA, pp. 139-152.
Sueoka, N. 1961. Compositional correlations between deoxyribonucleic acid and protein. Cold Spring Harb. Symp. Quant. Biol. 26, 35-43.
Sueoka, N. 1995. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J. Mol. Evol. 40, 318-325.
Szybalski, W., Kubinski, H., Sheldrick, P. 1966. Pyrimidine clusters on the transcribing strands of DNA and their possible role in the initiation of RNA synthesis. Cold Spring Harbor Symp. Quant. Biol. 31, 123-127.
Szybalski, W., Bovre, K., Fiandt, M., Guha, A., Hradecna, Z., Kumar, S., Lozeron, H. A., Maher, V. M., Nijkamp, H. J. J., Summers, W. C., Taylor, K. 1969. Transcriptional controls in developing bacteriophages. J.Cell Physiol. 74, suppliment 1, 33-70.
Tian, B., White, R. J., Xia, T., Welle, S., Turner, D. H., Mathews, M. B., Thornton, C. A. 2000. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA 6, 79-87.
Voinnet, O., Pinto, Y. M., Baulcombe, D. C. 1999. Suppression of gene silencing: a general strategy used by diverse DNA and RNA viruses of plants. Proc. Natl. Acad. Sci. USA. 96, 14147-14152.
Watson, J. D., Crick, F. H. C. 1953. Genetical implications of the structure of deoxyribonucleic acid. Nature 171, 964-967.
Winge, Ö. 1917. The chromosomes, their number and general importance. Compte. Rend. Trav. Lab. Carlsberg 13, 131-275.
![]()
For Chargaff's "In Praise of Smallness" (Click Here)
For Chargaff's "How Genetics Got a Chemical Education" (Click Here)
Go to: Bioinformatics Index (Click Here)
Go to: Homepage (Click Here)
![]()
This page was last edited on 04 Dec 2020 by Donald Forsdyke