|
|
D. R. Forsdyke
Journal of Molecular Evolution
(1995) 41, 573-581.
[With copyright permission from Springer. Received 18th October 1994. Accepted
25th April 1995]
Key words: Chargaff's rule - stem-loop - recombination - speciation.
A Note on Terminology not included in original paper: Just as A on one strand of a DNA duplex complements T on the other strand of the duplex (Chargaff's first parity rule), so, in natural DNA samples, A within a strand tends to match (numerically) T in the same strand (Chargaff's second parity rule). For a duplex the dinucleotide TC, for example, in the top strand has a complementary dinucleotide GA on the bottom strand. Note that since, by convention, we write DNA sequences from 5' to 3', and since the strands in a duplex are antiparallel (the 5' end of one strand is at the 3' end of the other strand), we write GA with the G on the bottom strand (which would base pair with the C on the top strand), written before the A (which would pair with the T in the top strand). Thus TC has as "complement" GA (or, when there is a need to distinguish, GA is the "reverse complement"), not AG (the "forward complement"). This also applies when we consider the frequencies of dinucleotides, trinucleotides, tetranucleotides, etc., in single stranded DNA. |
|
|
Hierarchy of Trinucleotide Frequencies
Relative Roles of Base Order and Base Composition
Fundamental Hierarchy of Dinucleotide Frequencies
Comparison of Trinucleotide Frequencies in Different Sequences
Base Composition and Base Order Determine the Frequencies of Complementary Trinucleotide Pairs
![]()
Summary. To an approximation Chargaff's rule (%A=%T; %G=%C) applies to single stranded DNA. In long sequences, not only complementary bases, but also complementary oligonucleotides are present in approximately equal frequencies. This applies to all species studied. However, species usually differ in base composition. With the goal of understanding the evolutionary forces involved, I have compared the frequencies of trinucleotides in long sequences and their shuffled counterparts. Among the 32 complementary trinucleotide pairs there is a hierarchy of frequencies which is influenced both by base composition (not affected by shuffling the order of the bases), and by base order (affected by shuffling). The influence of base order is greatest in DNA of 50% G+C, and seems to reflects a more fundamental hierarchy of dinucleotide frequences. Thus if TpA is at low frequency, all eight TpA-containing trinucleotides are at low frequency. Mammals and their viruses share similar hierarchies, with intra- and inter-genomic differences being mainly associated with differences in base composition (percentage G+C). E. coli and, to a lesser extent, Drosophila melanogaster hierarchies differ from mammalian hierarchies; this is associated with differences both in base composition and in base order. It is proposed that Chargaff's rule applies to single stranded DNA because there has been an evolutionary selection pressure favouring mutations that generate complementary oligonucleotides in close proximity, thus creating a potential to form stem-loops. These are dispersed throughout genomes and are rate-limiting in recombination. Differences in (G+C)% between species would impair interspecies recombination by interfering with stem-loop interactions. |
|
Introduction Chargaff (1951) observed in DNA that the percentage of A was equal to the percentage of T and the percentage of C was equal to the percentage of G. This symmetry "rule" lead to the A-T and G-C base pairing model of Watson and Crick (1953). Because it was concerned with pairing between the bases of opposite strands of the helix, the rule held precisely for both short and long DNA sequences. To a close approximation Chargaff's rule also applies to the base composition of single strands of a double helix, especially when long sequences (>1000 nt) are examined. It follows that, with very long sequences (e.g. 50,000 nt), there are equal proportions, not only of complementary single bases, but also of complementary dinucleotides, trinucleotides, tetranucleotides, etc. (Nussinov 1984; Alff-Steinberger 1984; Yomo and Ohno 1989; Prabhu 1993). While Chargaff's rule is species-independent, certain base ratios, (G+C)/(A+T), are species dependent (Chargaff, 1951). I here present studies of trinucleotide frequencies in natural and randomized (shuffled) DNA sequences of differing (G+C)%, with the goals of further understanding the biological relevance of the single strand symmetry of complementary oligonucleotides, the species-dependence of (G+C)%, and the evolutionary forces involved. |
|
Programs of the Genetics Computer Group (Gribskov and Devereux 1991) were made available on-line through the Molecular Biology Data Service of the National Research Council, Ottawa. Trinucleotide frequencies were obtained using the program COMPOSITION, which provides output as a square matrix of values for the 16 sets of 4 trinucleotides.
The middle bases of each set of 4 are G,A,T,C, in descending order. Members of the first row of 4 sets begin with G. Members of the second row begin with A. Members of the third row begin with T. Members of the fourth row begin with C. Members of the first column of 4 sets end with G. Members of the second column end with A. Members of the third column end with T. Members of the fourth column end with C. Pairs of complementary trinucleotides were selected by beginning at the top of the left column and proceeding down the columns from left to right.[Added note: see Table on right] Thus the first member of the top set of the left column was GGG. The value for this was assigned to the Y axis and was paired with the value of its complement CCC (X-axis), which was at the bottom of the bottom set of the fourth column. This assignment to axes was standard in plots of complementary trinucleotide frequencies. Sequences were randomized using the program SHUFFLE. Outputs from the latter could be used directly by COMPOSITION. Since COMPOSITION counts overlapping trinucleotides, the total number of trinucleotides in a sequence of N bases is N-2. |
|
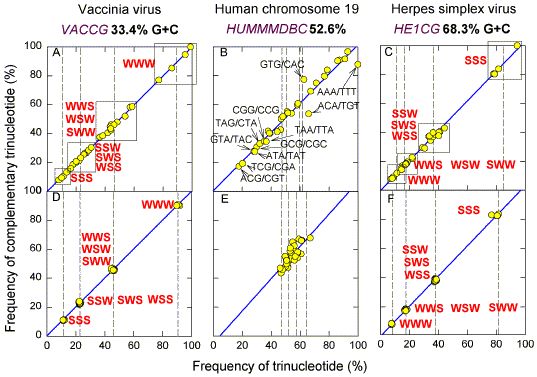
Hierarchy of Trinucleotide Frequencies The basic observation of Prabhu (1993) is shown in Fig. 1b for the case of the 32 pairs of complementary trinucleotides in the 68 kb GenBank sequence HUMMMDBC (Martin-Gallardo et al. 1992). The frequency of one member of each of the pairs is plotted relative to the Y-axis, and the frequency of the corresponding member relative to the X-axis. Frequencies are expressed as a percentage of the frequency of the most abundant trinucleotide (TTT for HUMMMDBC). Prabhu noted that members of complementary oligonucleotide pairs are present in similar frequencies, which usually differ from the frequencies of other pairs. Thus the experimental points are close to, and are widely distributed along, the diagonal. Prabhu made this observation with long DNA segments from many species, but did not examine the factors affecting the order of oligonucleotide frequencies and whether this order was specific for a particular DNA segment. |
|
|
| Fig. 1. Comparison of frequencies of
complementary trinucleotides in (A, B, C) the natural and (D, E, F) randomized versions of
one strand of (A, D) the vaccinia virus genome (GenBank; VACCG),
(B, E) a 68 kb human chromosome 19 segment (GenBank: HUMMMDBC),
and (C, F) the herpes simplex virus genome (GenBank: HE1CG).
Frequencies are expressed as percentages of the most abundant trinucleotide in the natural sequence. Each member of the 32 sets of trinucleotide pairs is assigned to either the X axis or the Y axis in a standardized way (see Methods; e.g. in the case of the point marked "GTG/CAC" the frequency of GTG refers to the Y coordinate, and the frequency of CAC refers to the X coordinate). "W" represents either A or T, and "S" represents either G or C. In (A) and (C) boxes surround points corresponding to the 8 members of the W3 group of trinucleotides (4 complementary pairs), the 24 members of the W2S group, the 24 members of the S2W group, and the 8 members of the S3 group. Vertical dashed lines indicate the average frequency among members of each group. The least-squares linear regression line for the 32 pairs of trinucleotides forms the diagonal (blue). |
|
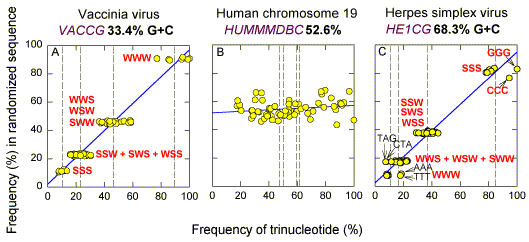
The dispersal along the diagonal is in keeping with studies showing a hierarchy of dinucleotide preferences in DNA sequences, which were attributed to "advantageous DNA structure" (Nussinov 1984). This is best understood in the cases of CpG and TpA. In many higher eukaryotes there has been a pressure to decrease the frequencies of the self-complementary dinucleotides CpG and TpA, and to increase the frequencies of the complementary dinucleotides TpG and CpA (Bird, 1980). There are eight trinucleotides containing the dinucleotide TpA, and eight trinucleotides containing the dinucleotide CpG. All 16 trinucleotides are of low frequency in HUMMMDBC (Fig. 1b). Thus, in this sequence changes in trinucleotide frequencies could be the result of changes brought about by evolutionary processes acting on the primary sequence at the dinucleotide level. Relative Roles of Base Order and Base Composition By shuffling the order of the bases it is possible to distinguish the roles of base composition- dependent factors from base order- dependent factors. When the order of bases in HUMMMDBC is randomized, the points corresponding to the frequencies of different trinucleotide pairs remain close to, but are much less widely distributed along, the diagonal (Fig. 1e). The difference between the dispersion along the diagonal seen in Figure 1b, and that seen in Figure 1e, provides a measure of the effect of base order on trinucleotide frequencies. In the case of HUMMMDBC, evolutionary forces appear to have influenced trinucleotide frequencies by affecting base order, rather than base composition. HUMMMDBC has a G+C content of 52.6%. Other GenBank sequences with G+C around 50% (HUMHDABCD, ECOUW89) give results similar to those in Figures 1b and 1e (except that CpG containing trinucleotides are abundant in the E. coli sequence ECOUW89). However, as base composition becomes progressively more distant from the 50% value, the general scatter of trinucleotide frequencies along the diagonal breaks down into four primary groupings. Using S to symbolize either G or C, and W to symbolize either A or T, the four groupings, in order, are (i) SSS (the "S3 group"; 4 pairs of complementary trinucleotides), (ii) SSW + SWS + WSS (the "S2W group"; 12 pairs of complementary trinucleotides), (iii) WWS + WSW + SWW (the "W2S group"; 12 pairs of complementary trinucleotides), and (iv) WWW (the "W3 group"; 4 pairs of complementary trinucleotides) [Added note 2000: distribution is according to the binomial theorum].This is shown well by two virus genomes with extreme G+C% values, vaccinia virus (Fig. 1a) and herpes simplex virus (Fig. 1c). In the case of vaccinia virus (33.4% G+C; Goebel et al. 1990), trinucleotides in the S3 group are least abundant and those in the W3 group most abundant. The converse applies in the case of herpes simplex virus (68.3% G+C; McGeoch et al. 1988). The groups are quite distinct from each other in the case of vaccinia virus, but in the case of herpes simplex virus there is an overlap between the W2S and the W3 groups (see later).When the sequences are randomized the dispersal of the trinucleotides of each group along the diagonal is greatly decreased (Figs 1d,f). Members of each group are clustered at four distinct positions. The mean frequency value for each trinucleotide group from the shuffled sequences (marked by vertical dashed lines in Figs. 1d,e,f), is similar to that for the corresponding group in the natural sequence (marked by vertical dashed lines in Figs. 1a,b,c). As DNA segments depart from 50% G+C, there is an increasing influence of base composition on trinucleotide frequencies. The influence is not absolute, since natural sequences show much more dispersion around the mean than randomized sequences. Even at extremes of (G+C)%, base order has some influence on trinucleotide frequency. Fundamental Hierarchy of Dinucleotide Frequencies The influence of base order in determining trinucleotide frequencies is particularly apparent in the case of TpA and CpG-containing trinucleotides. This is well shown when the frequencies of each of the 64 trinucleotides in a randomized sequence are plotted against their frequencies in the corresponding natural sequence (Fig. 2; Yomo and Ohno 1989). Although base composition is dominant, it is seen in the case of herpes simplex virus that a TpA-containing complementary pair from the W2S group (TAG/CTA) has a frequency corresponding to that of the W3 group. [It is also noted that a complementary pair from the W3 group (AAA/TTT) has a frequency corresponding to that of the W2S group.] |
|
|
| Fig. 2. Comparison of frequencies of the 64 trinucleotides in randomized ("shuffled") sequences with their frequencies in the corresponding natural, unshuffled, sequences. Trinucleotide frequencies, expressed as a percentage of the most abundant trinucleotide in the natural sequence, are plotted against each other. Other details are as in Fig. 1. |
|
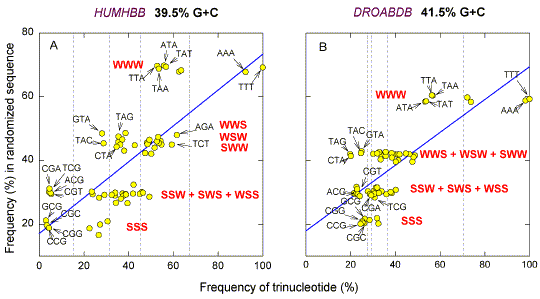
The effects of TpA and CpG are more evident in the case of sequences which deviate less extremely from 50% G+C. Fig. 3a shows a plot similar to Fig. 2 for the 73 kb human chromosome 11 segment containing the b-globin gene (HUMHBB; Lawn et al. 1980). Members of each trinucleotide group are more dispersed than in the case of the two viral genomes shown in Fig. 2. The frequencies of the four TpA-containing trinucleotides in each of the W3 and W2S groups are lower than the mean for the groups. Similarly the frequencies of the four CpG-containing trinucleotides in each of the S2W and S3 groups are lower than the mean for the groups. Similar results are observed for the 80 kb fruit fly segment DROABDB (Fig. 3b), and for the 315 kb sequence of the third chromosome of Saccharomyces cerevisiae (38.5% G+C; GenBank name SCCHRIII). In the latter case, the four CpG-containing members of the S3 group (SCG, CGS) occur, on average, 1836 times (standard error 22), while the four non-CpG-containing members occur, on average 234991 times. The four CpG-containing members of the S2W group (WCG, CGW) occur, on average, 286159 times, while the twenty other members occur, on average, 3796103 times. |
|
|
| Fig. 3. Comparison of frequencies of the 64 trinucleotides in randomized sequences with their frequencies in the corresponding natural sequences. Other details are as in Figs. 1 and 2. |
| The usual explanation for CpG
depletion is that the CpG sequence is a site of methylation in the germ
line and that 5-methylcytosine mutates by deamination to thymidine (Bird 1980). However, Drosophila
melanogaster and Saccharomyces cerevisiae DNAs are reported to contain no
detectable 5-methylcytosine (Urieli-Shoval et al. 1981; Proffitt et al. 1984). This
paradox will not be addressed here [Added note 2000: DNA of Drosophila
melanogaster contains 5-methylcytosine. Gowher et al., EMBOJ 19,
6918-6923].
The results shown in Figures 2 and 3 are in keeping with the proposal of Nussinov (1981) that dinucleotide frequencies are more fundamental than trinucleotide frequencies, so that we should seek to explain the latter in terms of the former, and not vice versa. |
|
|
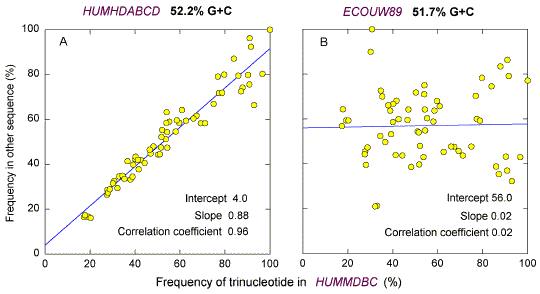
| Fig. 4. Comparison of frequencies of 64 trinucleotides in different sequences with G+C percentages around 50%. Trinucleotide frequencies in either (A) a 59 kb segment from human chromosome 4 (GenBank; HUMHDABCD), or (B) a 176 kb segment from E. coli (GenBank; ECOUW89), were plotted against the corresponding trinucleotide frequencies in the 68 kb segment from human chromosome 19 (GenBank; HUMMMDBC). The results of least-squares linear regression analysis are tabulated. |
|
Comparison of Trinucleotide Frequencies in Different Sequences Figure 1 has demonstrated a hierarchy of the frequencies of complementary trinucleotide pairs. The consistency of the order is evident in the two viral genomes where (G + C)% dominates; the order in the low (G+C)% genome (Fig. 1a) is the reverse of that in the high (G+C)% genome (Fig. 1c). Is the hierarchy consistent in DNA segments with around 50% G+C where effects of base order are most evident? Fig. 4a shows a plot of frequencies of the 64 trinucleotides in the 59 kb GenBank sequence HUMHDABCD (52.2% G+C; located on chromosome 4) against the frequencies of the corresponding 64 trinucleotides in the 68 kb GenBank sequence HUMMMDBC (52.6% G+C; located on chromosome 19). The rectilinear relationship shows that the hierarchical order of trinucleotide frequencies can be similar in widely separate human genome segments of similar G+C percentages. However, no such relationship is evident in the case of a 176 kb sequence from E. coli (ECOUW89), which has a similar percentage G+C (51.7%; Fig. 4b). There is no discernable correlation between the frequencies in the human DNA segment and the E. coli segment. Genomic segments from a variety of species were compared in a similar fashion. The results are shown in Table 1 where the sequences are ordered according to (G+C)%. The three linear regression parameters shown in Figure 4 are tabulated for each combination of segments. A perfect match, corresponding to the diagonal in the table, would have a Y-axis intercept at the origin (0), and slope and correlation coefficient (r) values of 1.0 (i.e. 0/1/1). With the exception of an E. coli segment (ECOUW89) and, to a lesser extent, a Drosophila segment (DROABDB), there is a general tendency for values to approach those of a perfect match as they approach the diagonal, either from the left or the right, or from above or below. Thus, the row corresponding to the vaccinia virus genome (VACCG) begins with a significant negative correlation (slope = -0.81). The slope then becomes progressively less negative and then becomes increasingly positive as the diagonal is approached. |
| Table 1. Summary of Linear Regression Data for Comparison of Trinucleotide Frequencies in Pairs of DNA Segments | ||||||||||||||
| GenBank DNA Segment |
Base Composition (% GC) |
GenBank DNA Segment (name abbreviated) | ||||||||||||
| . | HS4B9 |
HUMMM |
HUMHD |
ECOUW |
HUMGH |
MUSTC |
HUMTC |
RATCR |
DROAB |
HUMHP |
HUMHB |
HUMNE |
VACCG |
|
HS4B958RAJ |
60.0 | . | 5.6 |
6.5 |
35.2 |
16.3 |
31.9 |
33.0 |
36.5 |
52.6 |
43.6 |
46.1 |
47.5 |
58.2 |
HUMMMDBC |
52.6 | 20.3 |
. | -0.5 |
52.8 |
5.8 |
15.8 |
17.9 |
18.9 |
47.0 |
33.3 |
34.9 |
40.0 |
61.9 |
HUMHDABCD |
52.2 | 22.9 |
4.0 |
. | 41.4 |
8.9 |
17.1 |
18.8 |
19.7 |
43.5 |
30.6 |
33.2 |
37.4 |
58.4 |
ECOUW89 |
51.7 |
55.1 |
56.0 |
51.1 |
. | 58.8 |
61.7 |
59.1 |
62.8 |
47.4 |
57.6 |
59.3 |
57.4 |
59.5 |
HUMGHCSA |
49.2 |
33.4 |
5.9 |
5.1 |
62.9 |
. | 6.1 |
8.6 |
8.6 |
44.0 |
29.7 |
26.4 |
35.2 |
58.0 |
MUSTCRA |
44.9 |
43.5 |
14.0 |
12.8 |
59.6 |
4.8 |
. | 2.8 |
-0.08 |
27.3 |
17.0 |
13.3 |
20.9 |
38.8 |
HUMTCR- |
44.2 |
38.9 |
11.7 |
10.5 |
48.5 |
3.2 |
0.8 |
. | -1.4 |
19.1 |
12.5 |
9.2 |
15.7 |
31.7 |
RATCRYG |
43.8 | 48.0 |
19.7 |
18.5 |
60.7 |
10.6 |
4.1 |
6.2 |
. | 23.2 |
15.3 |
12.7 |
19.3 |
34.7 |
DROABDB |
41.5 |
47.4 |
31.0 |
29.4 |
21.8 |
26.5 |
18.2 |
15.3 |
12.9 |
. | 11.0 |
11.5 |
12.0 |
17.1 |
HUMHPRTB |
40.3 |
34.5 |
13.3 |
10.7 |
31.8 |
8.8 |
1.76 |
1.5 |
-2.5 |
2.9 |
. | 1.41 |
3.9 |
14.0 |
HUMHBB |
39.5 |
46.4 |
18.8 |
17.0 |
44.1 |
8.6 |
-1.3 |
-1.3 |
-5.7 |
4.9 |
2.3 |
. | 5.4 |
16.9 |
HUMNEUROF |
37.7 | 41.1 |
16.6 |
14.3 |
33.1 |
9.0 |
-1.2 |
-2.0 |
6.2 |
-2.6 |
2.8 |
-2.8 |
. | 10.2 |
VACCG |
33.4 | 70.4 |
50.0 |
50.4 |
47.2 |
37.2 |
18.2 |
15.1 |
8.8 |
-4.1 |
4.9 |
2.9 |
4.6 |
. |
| For
each segment pair the 64 trinucleotide
frequency values for a segment named in the left column were assigned to the Y axis and
the corresponding 64 values for the segment
named in the top row were assigned to the X axis. The three linear regression parameters
listed for each segment pair are, as in Fig. 4, the intercept at the Y axis, the slope and
the correlation coefficient (r). Note that only correlation coefficients
are independent of which segment of a pair is assigned to a particular axis. DNA segment sources are as follows: HS4B958RAJ, Epstein-Barr virus; HUMMMDBC, human chromosome 19 (Martin-Gallardo et al. 1992); HUMHDABCD, human chromosome 4; ECOUW89, E. coli; HUMGHCSA, human chromosome 17 region containing growth hormone genes; MUSTCRA, mouse chromosome containing T-cell receptor genes; HUMTCRADCV, human chromosome containing T-cell receptor genes; RATCRYG, rat chromosome containing g-crystallin gene cluster; DROABDB, Drosophila melanogaster; HUMHPRTB, human X chromosome region containing hypoxanthine-guanine phosphoribosyltransferase gene; HUMHBB, human chromosome 11 region containing the b-globin gene (Lawn et al. 1980); HUMNEUROF, human chromosome containing the gene implicated in neurofibromatosis; VACCG, complete genome of vaccinia virus (Goebel et al. 1990). |
||||||||||||||
|
These data show that the (G+C)%-associated ordering of the frequencies of complementary trinucleotide pairs forms a continuum extending across the chromosomes of one species (human), and across different mammalian species (rat, mouse), and includes some of their viruses (e.g. Epstein-Barr, HS4B958RAJ; Vaccinia, VACCG). Inter- and intragenomic differences in the hierarchy of trinucleotide frequences between mammalian species are mainly due to differences in (G+C)%. This also applies to their viruses, whose DNA contains mainly protein-encoding sequences. At odds with this is the E. coli segment (ECOUW89), which also consists mainly of protein-encoding sequences and has the same (G+C)% as HUMMMDBC and HUMHDABC, but shows no correlation with most sequences. Relative to HUMMMDBC, ECOUW89 appears much like a randomized sequence (cf. Figs. 2b, 4b). Being of approximately 50% G+C, the frequency of complementary trinucleotide pairs in ECOUW89 would be influenced mostly by base order (Fig. 1b), which would show dinucleotide frequency biases different from those of the mammalian sequences (Nussinov 1984). The only DNA segment with which ECOUW89 shows any significant correlation is the Drosophila melanogaster segment, DROABDB (r= 0.26; P<0.05). Thus, the E. coli and Drosophila hierarchies differ from the hierarchies of mammals and their viruses due to differences both in base composition and in base order, the latter likely reflecting differences in fundamental dinucleotide hierarchies. If complementary oligonucleotides are closely localized in a single strand of DNA (e.g. NNNTCANNNTGANNN) there should be an increased local potential to form stem-loops. This has been assessed (Forsdyke 1995a) by comparing the minimum free energy of folding for a 200 nt "window" in a natural sequence with the mean minimum free energy of folding for ten shuffled versions of the window (Le and Maizel 1989). The difference between the two values ("FORS-D value") provides a measure of the potential of the base order alone to contribute to secondary structure in the region of the window. The average values for windows at various intervals along sequences are shown in Table 2. Most windows have positive FORS-D values indicating a genome-wide evolutionary pressure distributed along the primary sequence favouring stem-loop formation. [Added note: The sign of the FORS-D value depends upon which way the substraction is carried out between the total stem-loop potential (FONS) and the base order-dependent stem-loop potential (FORS-M). In later work the order of substraction was changed, so that these values would then be recorded as negative.] The average values for three long human DNA segments from different chromosomes are very similar, whereas average values for E. coli segments are more variable. The lowest average value is for a fruit fly segment and the highest is for bacteriophage lambda. |
Table 2. Average FORS-D values for 200 nucleotide windows in DNA segments from different species. |
|||||
DNA sequence (GenBank name)a |
Length |
Base composition (% G+C) |
Number of windows |
Interval between start of successive windows (nucleotides) |
Average FORS-Db (kcal/mol) |
HUMHBB HUMHDABCD HUMMMDBC |
73,326 58,864 68,505 |
39.5 52.2 52.6 |
15 12 69 |
5,000 5,000 1,000 |
4.49 1.343.95 1.624.37 0.09 |
| . | |||||
DROABDB |
80,423 |
41.5 |
17 |
5,000 |
1.941.07 |
| . | |||||
ECOUW82 ECOUW85U ECOUW87 ECOUW89 ECO110K |
136,254 91,408 96,484 176,196 111,401 |
51.3 52.4 51.1 51.7 52.6 |
14 19 20 18 24 |
10,000 5,000 5,000 10,000 5,000 |
2.941.58 4.351.38 5.131.29 2.510.88 3.851.37 |
| . | |||||
LAMCG |
48,502 |
49.9 |
10 |
5,000 |
6.951.73 |
|
|||||
|
The experimental shuffling of the order of bases in a sequence of given length and base composition disrupts information present in the primary sequence and generates a reference sequence of the same length and base composition against which the natural sequence can be compared. Thus one can distinguish evolutionary pressures affecting base order from those affecting base composition. The data show that both natural DNA sequences and sequences derived from them by randomization, contain complementary trinucleotides in approximately equal proportions (i.e. all points fall close to the diagonal in Fig. 1). The studies of Prabhu (1993) suggest that this generalization could also apply to tetranucleotides, pentanucleotides, etc., although he did not examine randomized sequences. Since the shuffling of the order of bases in a sequence affects neither base composition, nor the equality of complementary trinucleotide frequencies, it is concluded that the single-strand symmetry of trinucleotide frequencies is a base composition- dependent property of DNA. This follows from the fact that, to an approximation, Chargaff's symmetry rule applies to long single stranded biological sequences. Did evolutionary forces select for the Chargaff ratios in single DNA strands, with equality of complementary oligonucleotide frequencies being an automatic consequence? Alternatively, did evolutionary forces select for equality of complementary oligonucleotide frequencies, with the Chargaff ratios being an automatic consequence? Base Composition and Base Order Determine the Frequencies of Complementary Trinucleotide Pairs Within the constraints imposed by (G+C)%, which become greater as (G+C)% departs from 50%, the greater dispersal of the experimental points along the diagonal in natural sequences relative to their randomized counterparts (Fig. 1), implies that there has been an evolutionary selection pressure acting on the primary sequence favouring this dispersal. The hierarchy of frequencies of trinucleotide pairs is maintained in various mammals and their viruses and reflects both base order (dinucleotide preferences) and base composition (Figure 4a; Table 1). The hierarchies in the relatively compact (with few or no introns) genomes of mammalian viruses form part of the same continuum as the hierarchies in the genomes of their hosts (Table 1); this indicates that in protein-encoding regions usage of redundant codons is sufficient to maintain a high degree of compatibility with percentage G+C. E. coli and, to a lesser extent, Drosophila melanogaster would depart from the continuum, not because of differences in base composition, but because of differences in base order. Evolutionary Selection to Enhance Stem-Loop Formation Since Chargaff's rule implies complementary base pairing, the equality of the frequencies of complementary oligonucleotides suggests that under biological conditions supercoiled DNA might have the potential to depart from its classical B-form duplex structure and form stem-loop cruciform structures, provided complementary oligonucleotides are suitably located (Murchie et al. 1992). Studies with anti-cruciform antibodies, a relatively crude measure, suggest that stem-loops are widely dispersed in genomes and may vary with the physiological state of the cell (Ward et al. 1990). Recently, the sequences of long segments of DNA have become available, permitting more detailed studies of the distribution of stem-loop potential. These studies, supplemented by the data of Table 2, show that the potential to form local stem-loops is both extensive and widely dispersed in genomes from a variety of species (Forsdyke 1995a-c). Thus, it seems that an organism which has accepted point mutations which increase the probability of stem-loop formation (both in protein-coding and in non-protein-coding DNA), has usually had an evolutionary advantage over an organism which has not accept such mutations. Furthermore, a sequence might not have been able to maximize simultaneously both its stem-loop and its protein-encoding potentials. It has been proposed that the conflict was resolved by permitting regions encoding a protein to arise in dispersed segments (exons), interrupted by regions of high stem-loop potential (introns; Forsdyke, 1995a-c). In that the Drosophila melanogaster genome is compact, with only short intergenic regions and introns, this organism might be more dependent on usage of redundant codons. The even more compact intronless E. coli genome might be similarly constrained. We are left with the question as to the possible evolutionary advantage of genome-wide stem-loop potential? One answer is that interactions between stem-loops of homologous chromosomes play an important role in the homology search preceding recombination (Forsdyke 1995a). Avoidance of recombination with organisms which have deviated from its own genomic sequence (incipient and closely related species) is essential if an organism is to use recombination with organisms which have not deviated (members of its own species), as a means of maintaining the integrity of its own DNA (Bernstein and Bernstein, 1991). Species differences in (G+C)% (and hence differences in oligonucleotide hierarchies), would have arisen to impair stem-loop interactions, and hence impair recombination between species. Differences in (G+C)% would reflect
Thus, other species constitute a selective force driving a species towards a particular (G+C)%. Acknowledgements. I thank D. Back for assistance in computer configuration and D. Bray of Queen's University StatLab for advice. The work was supported by a grant from the Medical Research Council of Canada. Alff-Steinberger C (1984) Evidence for a coding pattern on the non-coding strand of the E. coli genome. Nucleic Acids Res 12:2235-2241 Bernstein C, Bernstein H (1991) Aging, Sex and DNA Repair. Academic Press, San Diego Bird AP (1980) DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res 8: 1499-1504 Chargaff E (1951) Structure and function of nucleic acids as cell constituents. Fed Proc 10: 654-659 Filipski J (1990) Evolution of DNA sequence. Contributions of mutational bias and selection to the origin of chromosomal compartments. Adv Mutagenesis Res 2:1-54 Forsdyke DR (1995a) A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol Biol Evol 12: 949-958. Forsdyke DR (1995b) Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. An application of FORS-D analysis. Mol Biol Evol 12: 1157-1165. Forsdyke DR (1996) Different biological species "broadcast" their DNAs at different (G+C)% "wavelengths". J Theor Biol 178: 405-417. Goebel SJ, Johnson GP, Perkus ME, Davis SW, Winslow JP, Paoletti E (1990) The complete DNA sequence of vaccinia virus. Virology 179: 247-266 Gribskov M, Devereux J (1991) Sequence Analysis Primer. Stockton Press, New York Lawn RM, Efstratiadis A, O'Connell C, Maniatis T (1980) The nucleotide sequence of the human b-globin gene. Cell 21: 647-651 Le S-Y, Maizel JV (1989) A method for assessing the statistical significance of RNA folding. J Theor Biol 138: 495-510 Martin-Gallardo A, McCombie WR, Gocayne JD, Fitzgerald MG, Wallace S, Lee BMB, Lamerdin J, Trapp S, Kelley JM, Liu L-I, Dubnick M, Johnston-Dow LA, Kerlavage AR, Jong P de, Carrano A, Fields C, Venter JC (1992) Automated DNA sequencing and analysis of 106 kilobases from human chromosome 19q13.3. Nature Genet 1: 34-39 McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P (1988) The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol 69: 1531-1574 Murchie AIH, Bowater R, Aboul-ela F, Lilley DMJ (1992) Helix opening transitions in supercoiled DNA. Biochem Biophys Acta 1131: 1-15 Nussinov R. (1981) Eukaryotic dinucleotide preference rules and their implications for degenerate codon usage. J Mol Biol 149: 125-131 Nussinov R. (1984) Strong doublet preferences in nucleotide sequences and DNA geometry. J. Mol. Evol 20: 111-119 Prabhu VV (1993) Symmetry observations in long nucleotide sequences. Nucleic Acids Res 21: 2797-2800 Proffitt JH, Davie JR, Swinton D, Hattman S (1984) 5-Methylcytosine is not detectable in Saccharomyces cerevisiae DNA. Mol Cell Biol 4: 985-988. Urieli-Shoval S, Gruenbaum Y, Sedat J, Razin A (1981) The absence of detectable methylated bases in Drosophila melanogaster DNA. FEBS Lett 146: 148-152 Ward GK, McKenzie R, Zannis-Hadjopoulos M, Price GB (1990) The dynamic distribution and quantification of DNA cruciforms in eukaryotic nuclei. Exp Cell Res 188: 235-246 Watson JD, Crick FHC (1953) Genetical implications of the structure of deoxyribonucleic acid. Nature 171: 964-967 Yomo T, Ohno S (1989) Concordant evolution of coding and non-coding regions of DNA made possible by the universal rule of TA/CG deficiency - TG/CT excess. Proc Natl Acad Sci USA 86: 8452-8456 |
| Why are
complementary DNA strands symmetric?
This is the title of a fine paper by Pierre-Franois Baisne, Steve Hampson and Pierre Baldi (Bioinformatics 18, 1021-1033), that elegantly supports the above conclusions. The paper first notes that, unlike the above paper: "The literature apparently takes first-order [i.e. single bases] strand symmetry for granted in the first place. The underlying assumption is that base-compositional symmetry results from single point mutations that equally affect complementary strands, as demonstrated in the case of simple models of DNA evolution (Lobry, 1995; Lobry and Lobry, 1999). In addition, high order symmetry [i.e. duplets, triplets, etc.] is widely considered, implicitly or explicitly, as the consequence of first-order symmetry." It is further noted that: "Two high-order symmetry mechanisms, however, have been suggested. Fickett et al. (1992) noted that strand inversion - resulting from recombination events in which fragments of complementary strands are swapped - could be an explanation. From a more speculative perspective, Forsdyke (1995a, b) suggested that the selection of stem-loop structures might be a primary source of symmetry. ... We here demonstrate the existence of genuine high-order symmetries that do no entirely result from lower-order ones, and invalidate explanations relying on a single mechanism - be it single-point mutation at the first order [i.e. the Sueoka-Lobry view] , or recombinations events resulting in strand inversion [i.e. the Fickett view]." The paper concludes: "We establish the universality and variability range of first-order [i.e. single bases] strand symmetry, as well as its higher-order [i.e. duplets, triplets, etc.] extensions, and demonstrate the existence of genuine high-order symmetric constaints. We show that ubiquitous reverse complement symmetry does not result from a single cause, such as point mutation or recombination, but rather emerges from the combined effects of a wide spectrum of mechanisms operating at multiple orders and length scales" |
| Introns
Obey 2nd Parity Rule Better than Exons
Table 1 above demonstrates the use of the correlation coefficient (r) to compare tuple frequencies within and between species, as shown for the specific examples in Figure 4. Using this approach, in a fine study of 5-tuple frequencies in the genomes of the worm C. elegans and the fruit fly Drosophila melanogaster, Bultrini, Pizzi, Guidice and Frontali showed that the second parity rule is followed more closely in intronic and intergenic DNA than in exonic DNA (Gene 304, 183-192). They conclude:
|
| As added
to the text of the above paper, the pattern of distribution of
3-tuples in a randomized sequence is in accord with the binomial theorem
and is largely a function of base composition. Xie & Hao (2002)
have generalized this for higher tuple values, which are best
explained on the basis of Poisson distributions.
Xie, H. & Hao, B. (2002) Proc IEEE Comput Soc Bioinform Conf. 1:31-42. Visualization of K-tuple distribution in procaryote complete genomes and their randomized counterparts. |
| Some
important early papers, unfortunately overlooked when the paper was
written, are Philips et al. 1987 and Rogerson (1989, 1991). Table 2 of
the latter is very close to Table 1 (above).
Philips, G. J., Arnold, J. & Ivarie, R. (1997) Mono- through hexanucleotide composition of the Escherichia coli genome: a Markov chain analysis. Nucleic Acids Research 15, 2611-2626. Rogerson, A. C. (1989) The sequence symmetry of the Escherichia coli chromosome appears to be independent of strand or function and may be evolutionarily conserved. Nucleic Acids Research 17, 5547-5563. Rogerson, A. C. (1991) There appear to be conserved constraints on the distribution of nucleotide sequences in cellular genomes. Journal of Molecular Evolution 32, 24-30. [This paper cites a fundamental Markov chain study: Blaisdell, B. E. (1986) A measure of the similarity of sets of sequences not requiring sequence alignment. Proc. Natl. Acad. Sci. USA 83, 5155-5159. DRF Jan 2010] |
S peciation and diversification go hand-in-hand. Researchers construct phylogenetic trees that may model this diversification. Organisms showing similar characters are placed close on the trees. Organisms showing different characters are placed at greater distances on the trees. Some characters turn out to be more useful for tree construction than others. Naturally, when nucleic acid sequences became available, researchers used these for tree construction. The closer two sequences, the closer were considered the corresponding organisms.Various methods for multiple alignments of long strings of bases were introduced. This facilitated the counting of the number of base differences between the two sequences. However, the approach was empirical and did not take into account the possibility that some aspects of sequences, rather than just long strings of bases, might better relate to the underlying evolutionary processes that caused the species to diverge in the first place. For example, when spoken languages begin to diverge there is first a difference in accents. In this case, lining up long texts would necessitate the inclusion of much redundant information. Some measure of accent differences might much more precisely display the possible relationship between the languages because such redundancies would be eliminated. This paper would predict that k-tuple differences, be they 1-tuples (i.e. base composition) or higher order tuples, should provide better trees than multiple alignments. In this light it is noted that Kush Yang and Liqing Zhang (2008) now conclude that: "Trees constructed from the k-tuple distance are more accurate than those [constructed] from other distances most [of the] time; when the divergence between underlying sequences is high, the tree accuracy could be twice or higher using the k-tuple distance than other estimators." Qi J., Wang B. and Hao B. L. 2004 Whole proteome prokaryote phylogeny without sequence alignment: a K-string composition approach. J. Mol. Evol. 58, 1-11.[DRF 2010] Yang, K & Zhang, L. (2008) Performance comparison between k-tuple distance and four model-based distances in phylogenetic tree reconstruction. Nucleic Acids Res. 36, e33. |
The above alignment-free method has now been implemented as the "average nucleotide identity" (ANI) by Richter and Rossell-Mra (2009) for whom tetranucleotides are the oligonucleotide of choice as a "genomic gold standard" for species definition:
Richter, M. & Rossell-Mra, R. (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106, 19126-31. Sempath, R. et al. (2007) Rapid identification of emerging infectious agents using PCR and electrospray ionization mass spectrometry. Ann. New York Acad. Sci. 1102, 109-120. |
The alignment-free methodology that brings out the importance of higher order k-mers in speciation has now been supported by a wealth of studies that are summarized in two 2019 papers in a Linnean Society Journal (1,2). Furthermore, a brilliant statistical analysis of human genomes by Aggarwala and Voight (2016) suggests k-mer optimality when k=7 (3). However, another study with a wider range of species, suggests optima at k=5 or k=6 (4). Aggarwala and Voight conclude:
1. Forsdyke DR (2019a) Success of alignment-free oligonucleotide (kmer) analysis confirms relative importance of genomes not genes in speciation. Biol J Linn Soc 128, 239-250. 2. Forsdyke DR (2019b) Hybrid sterility can only be primary when acting as a reproductive barrier for sympatric speciation. Biol J Linn Soc 128, 779-788. 3. Aggarwala V, Voight BF (2016) An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nature Genetics 48, 349-355. 4. Morozov AA (2017) k-mer distributions of amino acid sequences are optimized across the proteome. bioRxiv doi: http://dx.doi.org/10.1101/190280. |
![]()
Go to: Abstract of above paper, 1994 (Click Here)
Return to: Prabhu's Symmetry Principle (Click Here)
Return to: Bioinformatics (Genomics) Index (Click Here)
Return to: HomePage (Click Here)
This page was established circa 1998 and was last edited on 19 January 2021 by Donald Forsdyke