Donald R. Forsdyke
Journal of Theoretical Biology (2007) 248, 745-753
DOI: 10.1016/j.jtbi.2007.07.008
With permission of the copyright holder - Else vier Ltd.
![]()
3. Principles of secondary structure calculation1. Introduction
2. RNA and DNA folding
![]()
|
The stability of a folded single-stranded nucleic acid depends on the composition and order of its constituent bases and may be assessed by taking into account the pairing energies of its constituent dinucleotides. To assess the possible biological significance of a computed structure, Maizel and coworkers in the 1980s compared the energy of folding of a natural single-stranded RNA sequence with the energies of several versions of the same sequence produced by shuffling base order. However, in the 2000s many took as self-evident the view that shuffling at the mononucleotide level (single bases) was conceptual wrong and should be replaced by shuffling at the level of dinucleotides (retaining pairs of adjacent bases). Folding energies then became indistinguishable from those of corresponding shuffled sequences and doubt was cast on the importance of secondary structures. Nevertheless, some continued productively to employ the single base shuffling approach, the justification for which is the topic of this paper. Because
dinucleotide pairing energies are needed to calculate structure, it does
not follow that shuffling should not disrupt dinucleotides. Base
shuffling allows determination of the relative contributions of base
composition and base order to total folding energy. The potential for
secondary structure arises from pressures acting at both DNA and RNA
levels, and is abundant throughout genomes - with a probable primary
role in recombination. Within a gene the potential can often be
accommodated, and base order and composition work together (values have
the same negative sign) in contributing to total folding energy. But
sometimes protein-coding pressure on base order conflicts with the
pressure for secondary structure and the values have opposite signs.
Total folding energy can be deemed of potential biological significance
when the average of several readings is significantly less than zero. Keywords: Base composition; Base order; Base shuffling; Dinucleotide; Folding energetics; Gene identification; MicroRNAs; Non-coding RNAs; Recombination; Secondary structure |
|
Apart from their encoding of proteins, single stranded nucleic acids have numerous other roles that involve their adoption of higher order structures (Forsdyke, 2006). Interest in computational approaches to the determination of nucleic acid structure has increased with the recognition of an abundance of non-coding RNAs (ncRNAs) in genomes, other than the well characterized ribosomal and transfer RNAs (rRNAs and tRNAs). However, the quest to identify and to establish genomic locations for ncRNAs through their structures is hindered by serious conceptual problems. The folding of a nucleic acid is hierarchical and sequential - the primary sequence determines secondary structure, which, in turn, determines higher ordered structure (Tinoco and Bustamante, 1999). A computer method for displaying the potential of successive segments of single-stranded nucleic acids to fold into secondary structures was developed by Le and Maizel (1989) for RNA, and was extended to DNA (Forsdyke, 1995a, b; Heximer et al. 1996). The difference between the folding energy value of a natural segment, and the mean of the folding energy values of several versions of the same segment generated by randomly shuffling base order, produced a metric ("segment score" or "FORS-D value") that could be related to functional aspects of the segment. However, the growing use of the method (specifically a study by Seffens and Digby in 1999), was brought into question by Workman and Krogh (1999), who considered that shuffling at the mononucleotide level (single bases) was conceptually wrong and should be replaced by shuffling at the level of dinucleotides (retaining pairs of adjacent bases). The observation that this dinucleotide shuffling approach failed to demonstrate that tRNAs had folding energies distinguishable from their shuffled versions, was dismissed as revealing that "the method is not always sensitive enough to discriminate between random sequences and RNA with a known secondary structure." The view of Workman and Krogh won wide support (Rivas and Eddy, 2000; Katz and Burge, 2003; Clote et al., 2005). Yet the alleged Le-Maizel house-of-cards did not tumble down over night. Some continued productively employing the individual base shuffling approach (Le et al., 2001, 2002, 2003; Forsdyke, 2002; Xue and Forsdyke, 2003; Washietl and Hofacker, 2004; Zhang et al., 2005a, b). Indeed, "good results" with the "simple model" appeared to justify "neglect" of dinucleotide shuffling (Washietl et al., 2005). Others, while seeing "no clear solution to the dilemma," suggested relaxation of "the constraint that every dinucleotide count -- be preserved" (Babak et al., 2007). I here discuss various aspects of nucleic acid folding in the hope of shedding some light on the controversy. It seems that, although there are problems with the original formulation of Le and Maizel (1989), there are even more with that of Workman and Krogh (1999). Furthermore, few seem to have thought in terms of a folding pressure arising primarily at the DNA level rather than at the level of the RNA transcribed from that DNA. |
|
Transcribed RNA is synthesized sequentially beginning at the 5' end and terminating at the 3' end. In the crowded intracellular environment, with protein concentrations around 300 mg/ml, the folding of the 5' end of RNA should begin prior to the synthesis of (and hence without necessary reference to the structure of) the 3' end (Forsdyke, 2006). The final structure would then be partly determined by this sequential mode of synthesis and partly by interaction with other cellular components, including RNA chaperones (Cristofari and Darlix, 2002). Approaches to determining the final operational structure of an RNA have included the computer-assisted folding of the entire sequence, or of sequential sections of that sequence, the latter approach being more likely to reflect a sequential mode of assembly during transcription. That the structure was likely to be of biological importance was obvious in the case of some non-protein-coding RNAs (tRNAs, rRNAs), but was not obvious for mRNAs. Yet, the potential for structure of mRNAs and the possibility that this might require accommodation to their protein-coding role has long been recognized (Ball, 1973; Forsdyke and Mortimer, 2000), and has gathered much support (Meyer and Miklos, 2005; Shabalina et al., 2006). Segments of single-stranded DNA are potentially extrudable from duplex DNA, especially when it has been subjected to negative supercoiling (Murchie et al., 1992; Krueger et al., 2006) and contains palindrome-like repeats (McMurray, 1999; Kogo et al., 2007). That this DNA property is widely and abundantly distributed along molecules, and is of general occurrence, is suggested by (i) the approximate equifrequencies of complementary oligonucleotides (e.g. CAT and AUG) throughout the genomes of many species ("Chargaff's second parity rule;" Forsdyke and Mortimer, 2000), (ii) direct measurements of folding potential (Forsdyke, 1995c; Heximer et al., 1996), and (iii) association with recombination (Zhang et al., 2005a, b). The duplex strand "unpairing" model of Crick (1971) postulated that such extrusion would occur during meiotic recombination (Forsdyke, 2007a). Many genomic translocations involve recombinations between sequences of similar base composition and distinctive propensities for secondary structure (Gotter et al., 2004). The propensity for such secondary structure would be conserved if it bestowed advantages either at the level of the conventional phenotype (natural selection) or of the genome phenotype (physiological "reprotypic" selection; Forsdyke, 2001, 2006). If not disadvantageous, conservation could also occur by chance isolation in small founder populations (random drift). |
3. Principles of secondary structure calculation
|
Calculations of the secondary structures of single-stranded nucleic acids take into account the energetics both of the stems (which usually contribute to stability) and of various loops and bulges (which usually decrease stability). Despite many complexities such calculated structures have proved valuable guides to the corresponding higher ordered structures, and hence to potential biological functions (Zuker, 2000; Mathews, 2006). The distribution of folding potential along a nucleic acid may be evaluated by calculating stability values (folding free energies) for consecutive windows (segments of uniform length) along the sequence. As far as the nucleic acid is concerned, the values arrived at depend - indeed, can only depend - on what bases are present in a window (base composition) and how they are ordered (base order). Although each element of a secondary structure (stems, loops, bulges) has to be considered separately, happily, decomposing a window sequence into its constituent overlapping dinucleotides (for each of which the energetics of base pairing with its complement is known) usually suffices to determine stem energetics, and it is not necessary to take into account higher order oligonucleotides (Borer et al., 1974; SantaLucia et al., 1996). Thus a trinucleotide (3 bases) can be decomposed into two overlapping "nearest neighbour" dinucleotides (each of 2 bases that overlaps the other by one base). Similarly, a tetranucleotide (4 bases) can be decomposed into three overlapping dinucleotides, the central of which overlaps its two "nearest neighbour" dinucleotides on either side. With such dinucleotide-pairing values, a sequence can be recursively test-folded (dynamic programming) until arrival at a structure (or a small set of closely related structures) whose stability values cannot be improved upon. The free energy is expressed in negative kilocalories/mol - more negativity implying greater stability. Folding patterns that favour stems increase the negativity, whereas folding patterns that favour loops (especially loop length) increase the positivity. A given natural sequence in a window is a member, both of a set of one with respect to its base order, and of a larger set of hypothetical sequences with the same base composition but differing in base order. While base composition is a major factor contributing to the window's total folding energy, a contribution of base order is often evident. The total folding energy (negative kilocalories/mol) is the sum of the base composition-dependent contribution (negative kilocalories/mol) and the base order-dependent contribution (either positive or negative kilocalories/mol). The latter can be evaluated indirectly by subtracting the base composition-dependent component for a window from the total folding energy (referred to as the "folding of natural sequence" value; FONS value) of that window (Table 1). |
|
Table 1. Base composition- and base order-dependent components of the folding energya |
||||||
|
|
||||||
|
Folding Energy |
|
Calculated as Folding of: |
|
Abbreviation |
|
Le-Maizel terminology |
|
|
|
|
|
|
|
|
|
Total |
|
Natural Sequence |
|
FONS |
|
E |
|
|
|
|
|
|
|
|
|
Base composition-dependent component |
|
Randomized Sequence Mean |
|
FORS-M |
|
Er |
|
|
|
|
|
|
|
|
|
Base order-dependent component |
|
Randomized Sequence Difference |
|
FORS-D |
|
E - Er |
|
a Base composition-dependent and base order-dependent components summate as the total folding energy. Thus FONS = FORS-M + FORS-D. |
||||||
|
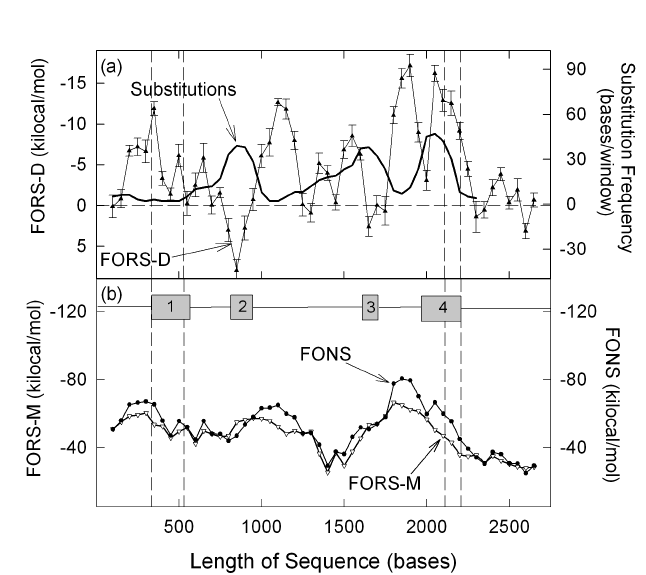
How is the base composition-dependent component determined? Some members of the set of hypothetical sequences with the same base composition as a natural sequence can be generated by randomizing (shuffling) the order of bases in the natural sequence. These members differ in base order, but have in common their base composition. Their individual folding energy values ("folding of randomized sequence" values; FORS values), reflect this commonality plus a contribution from base order that in some cases decreases the value (making them more negative), and in other cases increases the value (making them more positive). These individual idiosyncrasies are eliminated by taking the average of several members of the subset of shuffled sequences, to derive the "folding of randomized sequence mean" value (FORS-M value). This is the base composition-dependent component of the folding energy that is common to the natural sequence and all its shuffled versions. Since the FORS-M value is obtained by taking the mean of several FORS values that are normally distributed about the mean, it is accompanied by a value for the standard deviation that is affected by the size of the subset sample chosen for the calculation. When from a FONS value the FORS-M value is subtracted, one arrives at the "folding of randomized sequence difference" value (FORS-D value), which reflects the contribution of base order to the structure of the natural sequence. The latter value, together with the above standard deviation (or standard error), may be plotted against sequence position to display the distribution of FORS-D values in a sequence of interest. In most cases, a visual inspection suffices to reveal the statistical significance of FORS-D values at various positions along the sequence (Forsdyke, 1998). Alternatively, there is a metric to compare the score of one segment relative to the scores of all others in the sequence (Le et al., 1991). Since base composition tends to be more uniformly distributed, it is the base order-dependent component of the folding energy that can usually best be related to functional elements in a sequence. This approach can be applied to single-stranded sequences of RNA or DNA. The thermodynamics of base-pairing of dinucleotides with their complements (e.g. GU with AC, or GT with AC) differ for RNA and DNA, and different tables of dinucleotide pairing values are employed when computing folding energies (Nielsen et al., 1995; SantaLucia et al., 1996). However, only tables for RNA were available when I began my studies. When comparing successive windows in a DNA sequence I considered that these would suffice for observing broad qualitative differences (Forsdyke, 1995a); this was confirmed by later results obtained using tables for DNA when they became available (Heximer et al. 1996; Forsdyke, 1998). It was found that often negative FORS-D values supported negative FORS-M values to produce total negative folding values (FONS). However, in certain regions, especially exons, the signs of the FORS-D values changed (Forsdyke, 1995a). This was interpreted as revealing that when base order was responding to other pressures, such as the local exon-specific pressures for protein-encoding and purine-loading, then it might not be able to sustain its contribution to the total folding energy (Forsdyke and Mortimer, 2000). In other words, there was a conflict that might be resolved in favour of the other pressures. This view was supported by the observations that the conflict appeared greater in the cases of exons (i) that overlapped (thus being under greater protein-encoding and purine-loading pressures; Barrette et al., 2001), and (ii) that were deemed (on other grounds) to be under positive Darwinian selection (i. e. the protein sequence was evolving rapidly; Fig. 1; Forsdyke, 1995b; 1996a, b; 2007b). |
|
|
Fig.
1. Inverse relationship
between substitution frequency and base order-dependent stem-loop potential (FORS-D)
in exons of a gene under strong positive Darwinian selection (the rattlesnake
gene encoding the basic subunit of venom phospholipase A2). The
distribution of base substitutions (continuous line in (a)) is compared with
values for FORS-D (closed triangles, with standard errors of the mean), FORS-M
(open triangles), and FONS (closed circles). Values were determined for
overlapping 200 base windows that were moved in steps of 50 bases. Substitutions
are base differences relative to the rattlesnake acid subunit of venom
phospholipase A2. Grey boxes in (b) indicate the locations of the
four exons, with vertical dashed lines showing, consecutively, the beginning of
exon 1, the beginning of the protein-coding part of exon 1, the end of the
protein coding part of exon 4, and the end of exon 4. Adapted from Forsdyke
1995b ( |
|
The power of the moving window approach, where a window could be compared with its neighbours, was demonstrated by showing that the parts of an exon that were evolving most rapidly (in response to pressures at the protein level) had the most impairment of their base order-dependent folding potential (Forsdyke 1996a). Having confirmed this relative impairment of potential for secondary structures in exons, Babak et al. (2007) concluded that: "our analysis is not -- biased towards falsely reported structures, since some genomic features [e.g. introns] appear to be enriched for structures while others [e.g. exons] are depleted or neutral." Thus, "it seems unlikely that we are dramatically overestimating the degree of secondary structure in the genome because at least one type of genomic feature scores as containing less than would be expected by chance." So "there is likely to be an evolutionary selection against formation of secondary structure in open reading frames." The contribution of base order to stem-loop potential was found to be densely distributed throughout the genomes of a wide variety of species (Forsdyke, 1995c). There were also indications from a 68 kilobase DNA segment containing the human FOSB gene (chromosome 19) that plots of FORS-D values could assist gene identification in uncharted sequences (Heximer et al., 1996). Base order-dependent stem-loop potential could be compromised in exons, but this depended to the degree of conflict with other pressures (resulting in either negative or positive FORS-D values), so the general reliability of this method for exon identification was doubtful. Indeed, in a study of 51 entire mRNA sequences (i.e. not divided into windows) from a wide range of predominantly eukaryotic organisms (i.e. sequences representative of single or combined exon sequences), Seffens and Digby (1999) found 40 had negative FORS-D values, some very markedly so, while 11 had positive FORS-D values, albeit less marked. Similarly, of 125 entire human mRNA sequences, Jia and Luo (2006) found 103 had negative FORS-D values and 22 had positive FORS-D values, while of 107 entire E. coli mRNA sequences, 95 had negative FORS-D values and 12 had positive FORS-D values. Whether those with positive FORS-D values corresponded to genes under positive Darwinian selection, or overlapped other genes, was not stated in these studies. |
4. Base order as a measure of statistical significance
|
My studies (above) were well advanced when the pioneering work of Le and Maizel (1989) was noted. Without an awareness of the latter, the direction of the subtraction between FONS and FORS-M had initially been the reverse of that given above, so in early papers enhanced base order-dependent potential registered as positive (Forsdyke, 1995 a-d; 1996 a, b). This was corrected in Heximer et al. (1996) and in later papers, so that when FORS-M and FORS-D were acting in the same direction their signs were the same. However, some later contributors to the field (see below), reintroduced the incorrect method of subtraction, so a base-order dependent contribution to the stability of secondary structure was given a positive sign (Rivas and Eddy, 2000; Katz and Burge, 2003). There were substantial differences between my studies and those of the Maizel school (Table 1). Not only were there differences in direction of subtraction and nomenclature (e.g. FONS = "E"), but also in conceptual approach. Le and Maizel (1989) deemed the folding in a sequence window to be "statistically significant" if the base order-dependent component (FORS-D or "E - Er") was significantly different from the base composition-dependent component (FORS-M or "Er"). From this it could easily be inferred that the latter - the component usually making the largest contribution to the total folding energy - was somehow biologically insignificant. Similarly, Schultes et al. (1999) considered the base order-dependent component (FORS-D) to represent an aspect of a sequence that had "evolved," from which it could be inferred that the base composition-dependent component (FORS-M) had somehow not evolved. Noting that the "measure of minimum free energy is strongly correlated to the nucleotide composition" (assessed as the percentage of the bases G + C; i.e. base composition), Freyhult et al. (2005) incorrectly concluded that low (i.e. highly negative) FONS values do "not necessarily imply a stable structure." Irrespective of the properties of hypothetical sequences produced by shuffling, if a natural nucleic acid segment has a FONS value significantly less than zero (which is determined by repeated determinations of that FONS value), then its folding can be either biologically significant (functioning such that organisms without the folding might be eliminated by natural selection) or biologically insignificant (as when folding to a non-harmful level could have arisen by random drift in a population that was initially reproductively isolated in some way). The comparison of the folding of a natural sequence (FONS or "E") with the mean of multiple shuffled versions (FORS-M of "Er") could only determine the "statistical significance" of the extent to which base order served the folding function. Obtaining a value for FORS-D required a FORS-M yardstick, so a subset of a hypothetical set of sequences of the same base composition had first to be conjured up by shuffling the natural sequence. Against this yardstick it could be determined if base order had added to, substracted from, or made no difference to, the natural propensity of a sequence of that base composition to fold with a certain degree of stability (Forsdyke, 1998). Rather than expressing FORS-D values directly, Le and Maizel (1989) expressed "statistical significance" as the "segment score," namely the number of standard deviations by which the predicted free energy of the natural sequence was lower (more negative) than the average of the random sequences. The segment score (known in statistics as the Z-score), was calculated by dividing FORS-D ("E - Er") by the standard deviation (which depended on the size of the theoretical subset sampled). As applied to known RNA sequences, the approach revealed contributions of base order to structures that had been previously identified by other methods. An important outcome (although not acknowledged as such) arose when seeking a quick way to determine FORS-M values ("Er"). The contributions of several base composition-related parameters to FORS-M values were systematically investigated (Chen et al. 1990). Of these the product of the most strongly Watson-Crick pairing bases (G and C) proved of overwhelming importance as a predictor of the base composition-determined component of the folding energy. This was later independently confirmed (Forsdyke, 1998; 2007a). A quicker way of determining FORS-M values is now available (Washietl et al., 2005). That calculated structures (FONS values) were of biological significance was strongly suggested by demonstration of positive correlations with sequence conservation. Lawrence et al. (1991) showed that a conserved region in the bacterial ompA gene had a FONS value (-33.6 kcal/mol) that was lower than that of neighbouring regions, and was significantly lower than the corresponding FORS-M value (-27.3 kcal.mol), implying a contribution by base order of -6.3 kcal/mol (FORS-D value). It was inferred that it was nucleic acid structure, rather than amino acid sequence, that had been conserved (indicating a bias in choice among synonymous codons). This view is supported by recent studies covering a wide range of eukaryotic species that correlate structural constraints with usage of rare codons (Meyer and Miklos, 2005), and the demonstration for 20,000 human mRNAs that 5', coding and 3' domains tend to fold independently, with preferential base-pairing in coding domains occurring between the degenerate third position bases in codons (Shabalina et al., 2006). A relationship between conserved motifs of known function and FORS-D values was reported for the RNA genomes of various RNA viruses (Le and Maizel, 1989). For HIV-1 the relationship was highly significant for FORS-D, but not for FORS-M (Forsdyke, 1995d), so in this case it was conservation of base order rather than of composition that was critical. On the other hand, for the human FOSB gene (less likely to have been under positive Darwinian selection pressure) conservation correlated best with FORS-M values (Heximer et al., 1996). It should be noted that some transcribable elements of genomes may exist, not because they are conserved, but because they vary between individuals. Variation itself can be functional (Forsdyke et al., 2002; Pang et al., 2006). Thus, that which is evolutionarily conserved is often functional, but that which is functional is not necessarily conserved (Forsdyke, 2006). |
|
In the above, base order was disrupted absolutely by shuffling individual bases in an entire sequence window in order to obtain a FORS-M value for that window. In this way each of the natural dinucleotides (containing two consecutive bases) was replaced by a dinucleotide generated by the chance approximation of two bases during the shuffling process. Any compromise in the degree of shuffling would have decreased the disruption of the natural dinucleotides used for calculation of structure, and so would be expected to have decreased the difference from the FONS value for that window. Thus, values for the base order-dependent component, FORS-D, would have been compromised. The "statistical significance" of Le and Maizel (assessed as the "segment score" or Z-value) would have decreased. For example, when examining the central section of B. subtilis mRNAs, Rocha et al. (1999) obtained average FONS values of -6.9 kcal/mol and average FORS-M values of -6.1 kcal/mol, giving average FORS-D values of -0.8 kcal/mol. The latter fell to -0.3 kcal/mol when shuffling was limited to sustain trinucleotide compositions. Furthermore, as part of their study of entire mRNAs using the single base shuffle approach (see above), Seffens and Digby (1999) examined various ways of compromising base order less than absolutely (without changing base composition, but otherwise using the Le-Maizel approach). When only the protein-coding segments of the mRNAs were shuffled, the average segment score fell from -1.23 to -0.87, whereas when the protein-coding segments were left intact, but the non-coding segments were shuffled, the score fell to -0.50. It was deduced that both segments (coding and non-coding) contributed to the segment score (i.e. both segments contributed to the base order-dependent component of mRNA secondary structure). As expected, base order was compromised much less by simply shuffling the codons, thus changing the encoded protein, but disrupting (at best) only a third of the fundamental dinucleotide units that formed the basis of the structure calculations (i. e. the dinucleotide formed by the last base of one codon and the first base of its downstream codon was disrupted). This still resulted in a significant shift of average segment score from -1.23 to -0.63. Although segment score was affected by sample variance, which had decreased, the authors concluded that in a natural mRNA sequence the need for stable nucleic acid secondary structure would have influenced the sequence and hence codon choice. In other words, since the code was degenerate, in the course of the evolution of the sequence, codons which best served both protein-pressure and fold pressure had been selected (Itzkovitz and Alon, 2007). Since the energetics of pairing of complementary dinucleotides are major factors in calculations of folding energetics (see above), it would be predicted that if the dinucleotides (consecutive base pairs) rather than individual mononucleotides (single bases) of a natural sequence were shuffled, no dinucleotides would be interrupted and hence loss of base order would be minimized as far as the calculations were concerned. Thus, the differences between FONS and FORS-M values would be greatly decreased. This was observed by Workman and Krogh (1999) who studied both RNAs known to have distinctive structures related to their functions (5 tRNAs and 5 rRNAs), and a 46 member subset of the above set of mRNA sequences that had been studied by Seffens and Digby. When compared with their corresponding mononucleotide shuffled forms, 3 of the 5 tRNAs had negative Z-scores (implying negative FORS-D values), at least two of which were deemed significant. However, when compared with their corresponding dinucleotide shuffled forms, none were significant. Similarly, there was no evidence that natural mRNAs had lower folding free energies than sequences where the order of dinucleotides had been randomized (codon order not being retained), a result later confirmed by Clote et al. (2005). Nevertheless, Workman and Krogh confirmed the results of Seffens and Digby for shuffled single bases (mononucleotides): of the 46 sequences, 38 had negative Z-scores and 8 were positive (average Z score of -1.59). Intriguingly, in the case of the 5 rRNAs, whether shuffling was at the dinucleotide or mononucleotide level, all had significantly negative Z-scores, but 4 of the 5 had better (more negative) Z-scores when shuffling was performed at the mononucleotide level. Despite the small number of non-protein-coding RNAs studied, on balance the results of Workman and Krogh appeared consistent with the above prediction. But the authors went beyond this, implying that the single base shuffling approach, as used in all the studies cited earlier in this paper, was misleading; mRNAs in general do "not form more stable extended structures than random sequences." Rivas and Eddy (2000) took the matter further, declaring that the merely "anecdotal" work of the Maizel school was a "statistical artifact" that came "mostly from local base composition bias and not from RNA secondary structure." Katz and Burge (2003) thought that Workman and Krogh had "challenged" with "justified criticism" the notion of a "higher stability of RNA structures in native sequences," since account had not been taken of the fact that "RNA folding thermodynamics is strongly dependent on dinucleotide stacking interactions." Clote et al. (2005) agreed that for mRNA "valid conclusions" could not be drawn from the single base shuffling approach. Katz and Burge (2003) sought to resolve the "contradiction," referring to a normalized FORS-D values as "excess folding potential" (EFP) and reversing its sign to positive when the natural sequence was more stable (see above), but retained the negative convention for the Le-Maizel "segment scores" (Z-scores). An ingenious procedure ("dicodon shuffle") was introduced for partially disrupting base order in a bacterial mRNA sequence while preserving its dinucleotide composition, and its codon usage and order (i.e. the same protein sequence was encoded). With this approach significant differences from FONS values were claimed, indicating "widespread selection for local RNA secondary structure." However, whereas in most other studies sequences were either undivided or divided into windows of around 200 bases prior to shuffling and determining fold energy values, Katz and Burge shuffled entire mRNA sequences prior to dividing them into short 50 base windows for determining energy values. The windows that were shuffled and folded were not the same as the natural windows that were folded without shuffling. It is likely that the some of the shuffled windows would have differed in total base compositions, dinucleotides and synonymous codons, from the corresponding natural windows. In view of the great sensitivity of folding to small differences in base composition, full acceptance of their results must be deferred pending clarification. It is of note, however, that the studies of Shabalina et al. (2006) with eukaryotic mRNAs, while appearing consistent with those of Seffens and Digby (1999), resulted in sequences with the same folding energy as the natural sequence when the "dicodon shuffle" procedure was employed. Furthermore, when seeking to identify genes corresponding to non-protein-encoding RNAs, Babak et al. (2007) observed that "false positives rates drastically increased when using shuffles with conserved dinucleotides," compared with shuffles "without preservation of dinucleotide content." How did Workman and Krogh arrive at their position? They noted the well known fact that, just as genomes of different species often differ in base compositions (e.g. percentage G + C), they also often differ in dinucleotide (and higher oligonucleotide) compositions (Nussinov, 1984; Forsdyke, 1995c; Karlin et al., 1997). Thus, compared with an (A + T)-rich genome, a (G + C)-rich genome will tend to have a higher base composition-dependent contribution to stem-loop potential (Woodside et al., 2007). Such a genome will also have more dinucleotides that pair strongly with complementary dinucleotides (e.g. GC pairs more strongly with its complement, GC, than AT pairs with its complement, AT; Yakovchuk et al., 2006). They also noted that "the physical stability of RNA secondary structure is known to depend on dinucleotide base stacking energies." That these facts should justify dinucleotide shuffling and prohibit mononucleotide shuffling was deemed self-evident - no further explanation was offered. Matters were compounded by the statement that "it is unlikely that the dinucleotide distribution of mRNA is influenced by the need to form secondary structure because the dinucleotide distribution is generally very similar in other types of DNA from the same organism (i.e. non-coding DNA)." It would appear from this that Workman and Krogh were unaware of the extensive studies (outlined above) showing that the potential for secondary structure is widely distributed in non-genic DNA (Forsdyke, 1995a, b, c; 1996a, b; 1998). This may partly explain why Rivas and Eddy (2000) concluded from a preliminary survey using the Le-Maizel approach (mononucleotide shuffling) that it would be difficult to find structural signals allowing identification of genomic locations corresponding to non-protein-coding RNAs (i.e. RNAs that might function by virtue of their structure rather than as mRNAs). In thermophilic bacteria this task was found easier. Thermophilic bacteria maintain the stability of non-protein-coding RNA structures by increasing their G + C content. That mRNAs do not require similar degrees of structure was indicated by their G + C content being marginally decreased in thermophiles (Galtier and Lobry, 1997; Lambros et al., 2003). Hence the locations of some non-protein-coding RNAs in the genomes of thermophiles are detectable on the basis of G + C content alone (Schattner, 2002). This was found "surprising" by Klein, Misulovin and Eddy (2002). The DNA of the genes from which non-protein-coding RNAs are transcribed is likely to have the potential for extruded secondary structures that may function both in the nucleus (at the DNA level), and in the cytoplasm (at the RNA level; Forsdyke, 2006). Indeed, more extensive studies have shown that, in contrast to mRNAs, and as suggested for rRNAs by the preliminary studies of Workman and Krogh (1999), some non-protein-coding RNAs that function by virtue of their structures have significantly lower (more negative) FONS values than the mean value of the corresponding shuffled sequences, even when shuffling is at the dinucleotide level (Bonnet et al., 2004; Clote et al., 2005; Freyhult et al., 2005). The explanation for this is not clear at this time. However, studies with thermophiles provide the best evidence that mRNAs themselves do not require higher ordered structure, at least not to the same extent as non-protein-coding RNAs. Since most mRNAs still display considerable potential for such structure, this suggests that the primary pressure for their structure is at the level of the DNA from which they are transcribed. Mechanisms by which thermophiles, even when low in G + C, maintain the stability both of normal duplex DNA and of its extruded stem-loop structures, are considered elsewhere (Lambros et al., 2003). |
|
Procedures
for quantitating the potential of successive segments of single-stranded nucleic
acids to fold into secondary structures were independently introduced by the
Maizel school (RNA; terminology E, Er, and E - Er) and
Forsdyke (DNA; terminology FONS, FORS-M and FORS-D). In error, FORS-D was
initially calculated by subtracting FONS from FORS-M (i.e. Er - E),
instead of the converse. This was later corrected. However, conceptual
differences remained. The Maizel school saw E- Er (FORS-D) as a
measure of the
"statistical significance" of folding, whereas Forsdyke saw
it as a measure of the contribution of base order to the folding energy, to be
contrasted with the contribution of base composition, Er (FORS-M). Further
problems arose when it was postulated by Workman and Krogh that the shuffling
for determining Er should be at the level of dinucleotides. This
paper has made the following case. The
statistical significance of the calculated folding energy of a natural
single-stranded nucleic acid sequence is determined by repeating the folding
several times and showing that the average E value (FONS value) is significantly
different from zero. The fact that dinucleotide, rather than mononucleotide,
pairing energies are necessary for the initial folding energy calculations,
merely confirms that base composition and base order are both important
determinants of structure. To distinguish these determinants, shuffling is
performed at the mononucleotide level. Shuffling at the dinucleotide level does
not facilitate a decision on the significance of mRNA structure, but may be
telling us something about some other RNA types. Since duplex DNA sequences have
an extensive natural propensity to extrude single-strand structures, genomic
searches for transcribable elements on the basis of structural differences may
not be successful. Such elements are likely to be detected either by virtue of
conflicts with the natural propensity of duplex DNA for extruded structure (Heximer
et al., 1996), and/or by combining the latter thermodynamic measure with a
measure of sequence conservation (Washietl et al., 2005). However, conservation
may not be a sine qua non of such elements (Forsdyke et al., 2002). To the extent that these conclusions prove valid, then it is perhaps to the history of science that we must go for explanations. A century ago the dominations of the Darwinian natural selectionists and biometricians were challenged by the early geneticists struggling at the interface between disciplines (Provine, 1971; Forsdyke, 2001). It can be anticipated that the present dominations by the Darwinian genocentrists and informaticists will be increasingly challenged by the early evolutionary bioinformaticists whose interface struggles will be no less poignant (Forsdyke, 2006). |
REFERENCES
Babak, T., Blencowe, B. J., Hughes, T. R., 2007. Considerations in the identification of functional RNA structural elements in genomic alignments. BMC Bioinf. 8, 33.
Ball, L. A., 1973. Mutual influence of the secondary structure and information content of a messenger RNA. J. Theor. Biol. 41, 243-257.
Barrette,
Bonnet, E., Wuyts, J., Rouze, P., Peer, Y. V. de, 2004. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 20, 2911-2917.
Borer, P. N., Dengler, B., Tinoco,
Chen, J.-H., Le, S.-Y., Shapiro, B., Currey, K. M., Maizel, J. V., 1990. A
computational procedure for assessing the significance of RNA structure. CABIOS
6, 7-18.
Clote, P., Ferre, F., Kranakis, E., Krizanc, D., 2005. Structural RNA has a
lower folding energy than random RNA of the same dinucleotide frequency. RNA
11,
578-591.
Crick, F., (1971) General model
for the chromosomes of higher organisms. Nature
234, 25-27.
Cristofari, G., Darlix, J.-L., (2002) The ubiquitous nature of RNA chaperone
proteins. Prog. Nuc. Acid Res. Mol.
Biol. 72, 223-268.
Forsdyke, D. R., 1995a. A stem-loop "kissing" model for the initiation
of recombination and the origin of introns. Mol. Biol. Evol. 12,
949-958.
Forsdyke, D. R., 1995b. Conservation of stem-loop potential in introns of snake
venom phospholipase A2 genes. An application of FORS-D analysis. Mol.
Biol. Evol. 12, 1157-1165.
Forsdyke, D. R., 1995c. Relative roles of primary sequence and (G+C)% in
determining the hierarchy of frequencies of complementary trinucleotide pairs in
DNAs of different species. J. Mol. Evol. 41, 573-581.
Forsdyke, D. R., 1995d. Reciprocal relationship between stem-loop potential and
substitution density in retroviral quasispecies under positive Darwinian
selection. J. Mol. Evol. 41, 1022-1037.
Forsdyke, D. R., 1996a. Stem-loop potential in MHC genes: a new way of
evaluating positive Darwinian selection. Immunogenetics 43, 182-189.
Forsdyke, D. R., 1996b. Different biological species "broadcast" their
DNAs at different (C+G)% "wavelengths". J. Theor. Biol. 178,
405-417.
Forsdyke, D. R., 1998. An alternative way of thinking about stem-loops in DNA. A
case study of the human G0S2 gene. J. Theor. Biol. 192, 489-504.
Forsdyke, D. R., 2001. The Origin of Species, Revisited. McGill-Queen's University Press, Montreal.
Forsdyke, D. R., 2002. Selective
pressures that decrease synonymous mutations in Plasmodium falciparum.
Trends Parasitol. 18,
411-418.
Forsdyke, D. R., 2006. Evolutionary Bioinformatics. Springer,
New York.
Forsdyke, D. R., 2007a. Molecular sex: the importance of base composition rather
than homology when nucleic acids hybridize. J. Theor.
Biol. (in press) (Click here)
Forsdyke, D. R., 2007b. Positive Darwinian selection. Does the comparative
method rule? J. Biol. Sys. 15,
95-108.
Forsdyke, D. R., Madill, C. A., Smith, S. D., 2002. Immunity as a function of the unicellular state: implications of emerging genomic data. Trends Immunol. 23, 575-579.
Forsdyke, D. R., Mortimer, J. R.,
2000. Chargaff's legacy. Gene 261, 127-137.
Freyhult, E., Gardner, P. P., Moulton, V., 2005. A comparison of RNA folding
measures. BMC Bioinformatics 6, 241.
Galtier, N., Lobry, J. R., 1997.
Relationships between genomic G + C content, RNA secondary structures, and
optimal growth temperature in prokaryotes. J. Mol. Evol.
44, 632-636.
Gotter, A. L., Shaikh, T. H., Budarf, M. L., Rhodes,
C. H., Emanuel, B. S., 2004. A
palindrome-mediated mechanism distinguishes translocations involving LCR-B of
chromosome 22q11.2.
Hum. Mol.
Genet. 13, 103-115.
Heximer, S. P., Cristillo, A. D., Russell, L., Forsdyke, D. R., 1996. Sequence analysis and expression in cultured lymphocytes of the human FOSB gene (G0S3). DNA Cell Biol. 15, 1025-1038.
Itzkovitz, S., Alon, U., 2007. The genetic code is nearly optimal for allowing additional information within protein-coding sequences. Genome Res. 17, 405-412.
Jia, M., Luo, L., 2006. A relation between mRNA folding and protein structure. Biochem. Biophys. Res. Comm. 343, 177-182.
Karlin, S., Mrazek, J., Campbell, A. M., 1997. Compositional biases of bacterial genomes and evolutionary implications. J. Bact. 179, 3899-3913.
Katz, L., Burge, C. B., 2003.
Widespread selection for local RNA secondary structure in coding regions of
bacterial genes. Genome Res. 13, 2042-2051.
Klein, R. J., Misulovin, Z., Eddy, S. R., 2002. Noncoding RNA genes identified in AT-rich
hyperthermophiles. Proc. Natl. Acad. Sci. USA
99, 7542-7547.
Kogo, H., Inagaki, H., Ohye, T., Kato, T., Emanuel, B. S., Kurahashi, H., 2007.
Cruciform extrusion propensity of human translocation-mediating palindromic
AT-rich repeats. Nucleic Acids Res. 35, 1198-1208.
Krueger, A., Protozanova, E., Frank-Kamenetskii, M. D., 2006. Sequence-dependent basepair opening in DNA double helix. Biophys. J. 90, 3091-3099.
Lambros, R. J., Mortimer, J. R.,
Forsdyke, D. R., 2003. Optimum growth temperature and the base composition of
open reading frames in prokaryotes. Extremophiles
7, 443-450.
Lawrence, J. G., Hartl, D. L., Ochman, H., 1991. Molecular considerations in the
evolution of bacterial genes. J. Mol. Evol.
33, 241-250.
Le, S.-Y., Chen, J.-H., Konings,
D., Maizel, J. V., 2003. Discovering well-ordered folding patterns in nucleotide
sequences. Bioinformatics 19, 354-361.
Le, S.-Y., Chen, J.-H., Maizel, J. V., 1991. Detection of unusual RNA folding
regions in HIV and SIV
sequences. CABIOS 7, 51-55.
Le, S-Y., Liu, W-M., Chen, J-H., Maizel, J. V., 2001. Local thermodynamic stability scores are well represented by a non-central Student's t distribution. J. Theor. Biol. 210, 411-423.
Le, S.-Y., Maizel, J. V., 1989. A
method for assessing the statistical significance of RNA folding. J.
Theor. Biol. 138, 495-510.
Le, S.-Y., Zhang, K., Maizel, J. V., 2002. RNA molecules with structure
dependent functions are uniquely folded. Nucleic Acids
Res. 30, 3574-3582.
Mathews, D. H., 2006. Revolutions
in RNA secondary structure prediction. J. Mol. Biol.
359, 526-532.
McMurray, C. T., 1999. DNA secondary structure: A common and causative factor for expansion in
human disease. Proc. Nat. Acad. Sci. USA
96, 1823-1825.
Meyer, I. M., Miklos,
Murchie, A. I. H., Bowater, R., Aboul-Ela, F., Lilley, D. M. J., 1992. Helix
opening transitions in supercoiled DNA. Biochem. Biophys. Acta
1131,
1-15.
Nielsen, D. A., Novoradovsky, A., Goldman, D., 1995. SSCP primer design based on
single-stranded DNA structure predicted by a DNA folding program. Nucleic Acids
Res. 23, 2287-2291.
Nussinov, R. (1984) Doublet frequencies in evolutionarily distinct groups. Nucleic
Acids Res. 12, 1749-1763.
Pang, K. C., Frith, M. C., Mattick,
J. S., 2006. Rapid
evolution of noncoding RNAs: lack of conservation does not mean lack of function.
Trends Genet. 22, 1-5.
Provine, W. B., (1971) The Origins of Theoretical Population
Genetics. University
of
Rivas, E., Eddy, S. R., 2000. Secondary structure alone is generally not
statistically significant for the detection of non-coding RNAs. Bioinformatics
16, 583-605.
Rocha, E. P. C., Danchin, A., Viari, A., 1999. Translation in Bacillus
subtilis: role and trends of initiation and termination, insights from a
genome analysis. Nucleic Acids Res. 27, 3567-3576.
SantaLucia, J., Allawi, H. T., Seneviratne, P. A., 1996. Improved nearest-neighbor
parameters for predicting DNA duplex stability. Biochemistry
35, 3555-3562.
Schattner, P., 2002. Searching
for RNA genes using base-composition statistics. Nucleic Acids Res. 30, 2076-2082.
Schultes, E. A., Hraber, P. T., LaBean, T. H., 1999. Estimating the
contributions of selection and self-organization in RNA secondary structure. J.
Mol. Evol. 49, 76-83.
Seffens, W., Digby, D., 1999.
mRNAs have greater negative folding free energies than shuffled or codon-choice
randomized sequences. Nucleic
Acids Res. 27, 1578-1584.
Shabalina,
S. A., Ogurtsov, A. Y., Spiridonov, N. A., 2006. A periodic pattern of mRNA
secondary structure created by the genetic code. Nucleic Acids
Res. 34,
2428-2437.
Tinoco,
Washietl, S., Hofacker,
Washietl, S., Hofacker,
Woodside, M. T., Anthony, P. C., Behnke-Parks, W. M., Larizadeh, K., Hershlag, D., Block, S. M., 2007. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science 314, 1001-1004.
Workman, C., Krogh, A., 1999. No evidence that mRNAs have lower folding free energies than random sequences with the same dinucleotide distribution. Nucleic Acids Res. 27, 4816-4822.
Xue, H. Y., Forsdyke, D. R., 2003. Low complexity segments in Plasmodium falciparum proteins are primarily nucleic acid level adaptations. Mol. Biochem. Parasitol. 128, 21-32.
Yakovchuk, P., Protozanova, E., Frank-Kamenetskii, M. D., 2006. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 34, 564-574.
Zhang, C.-Y., Wei, J.-F., He, S.-H., 2005a. Local base order influences the origin of ccr5 deletions mediated by DNA slip replication. Biochem. Genet. 43, 229-237.
Zhang, C.-Y., Wei, J.-F., He,
S.-H., 2005b. The key role for local base order in the generation of multiple
forms of China HIV1 B'/C intersubtype recombinants. BMC Evol.
Biol. 5, 53.
Zuker, M., 2000. Calculating
nucleic acid secondary structure. Curr. Opin. Struct. Biol. 10, 303-310.
![]()
Spano and coworkers (2008), noting that several teams "have been productively employing mononucleotide shuffling in their research investigations," compared shuffling at the mononucleotide and dinucleotide levels (Z-scores) and elected to use mononucleotide shuffling to evaluate the structures of the corresponding natural sequences ("random null hypothesis"). From a study of 1212 complete virus genomes, they concluded:
Spano, M., Lillo, F., Micciche, S. & Mantegna, R. N. (2008) Statistical properties of thermodynamically predicted RNA secondary structures in viral genomes. Eur. Phys. J. B. 65, 323-331. |
Einert and coworkers (2011), note that a classical model for duplex melting - the Poland-Scheranga model - does not take into account the fact of widely dispersed stem-loop potential. While explicitly referring to RNA, Einert et al. "note that in principle all our results carry over to single-stranded DNA molecules as well."
One might add, important also for DNA recombination. They conclude that the Poland-Scheranga model only applies when stem-loop potential is absent: "In the case where intra- and inter-strand base pairing occurs, the classical Poland-Scheranga mechanism for the melting of a DNA duplex has to be augmented by the single-strand folding scenario considered by us, as the Poland-Scheranga theory is only valid in the case where no intra-strand base pairing is possible." Einert TR, Orland H & Netz RR (2011) Secondary structure formation of homopolymeric single-stranded nucleic acids including force and loop entropy: implications for DNA hybridization. Eur. Phys. J. E. 34, 55. |
![]()
Go to Bioinformatics Index Click Here
Go to Home Page Click Here
![]()
This page was established in July 2007 and was last edited 08 Nov 2020 by Donald Forsdyke