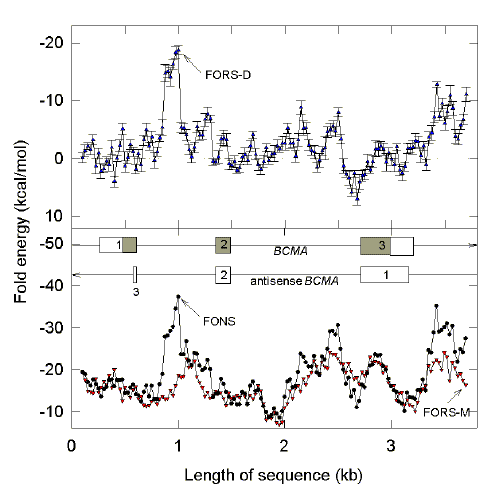
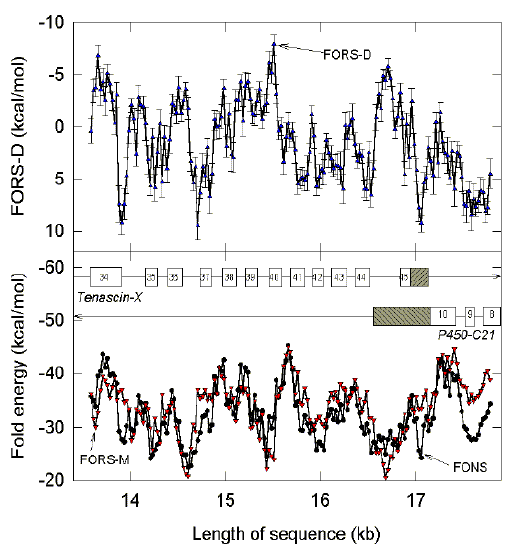
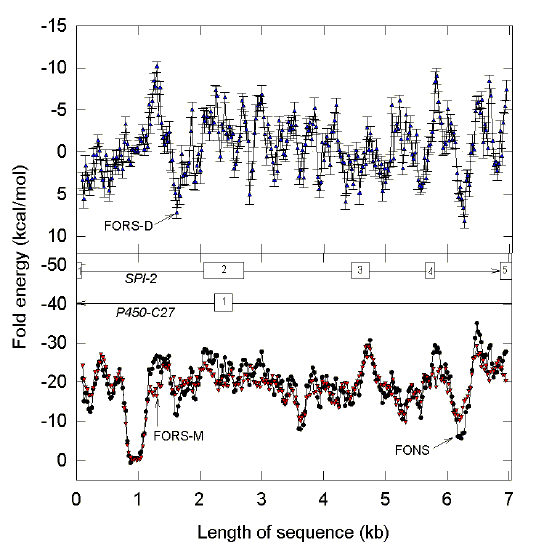
|
References
Alvarez-Valin, F., Tort, J. F., Bernardi, G. 2000. Nonrandom spatial
distribution of synonymous substitutions in the GP63 gene from Leishmania. Genetics
155,1683-1692.
Ball, L. A. 1973. Mutual influence of the secondary structure and information
content of a messenger RNA. J. Theor. Biol.
41, 243-247.
Bell, S. J., Forsdyke, D. R.1999. Deviations from Chargaff's second parity
rule correlate with direction of transcription. J.
Theor. Biol. 197, 63-76.
Bronson, E. C., Anderson, J. N. 1994. Nucleotide composition as a driving
force in the evolution of retroviruses. J. Mol.
Evol. 38, 506-532.
Carlson, E. A. 1981. Genes, Radiation and Society. The Life and Work of H. J.
Muller. Cornell University Press, Ithaca, pp. 390-392.
Crick, F. 1971. General model for the chromosomes of higher organisms. Nature
234, 25-27.
Doyle, G. G. 1978. A general theory of chromosome pairing based on the
palindromic model of Sobell with modifications and amplification. J.
Theor. Biol. 70,171-184.
Eguchi, Y., Itoh, T., Tomizawa, J. 1991. Antisense RNA. Annu. Rev.
Biochem. 60, 631-652.
Forsdyke, D. R. 1981. Are Introns In-series Error-detecting Sequences? J.
Theor. Biol. 93, 861-866.
Forsdyke, D. R. 1995a. A stem-loop "kissing" model for the
initiation of recombination and the origin of introns. Mol. Biol. Evol.
12, 949-958.
Forsdyke, D. R. 1995b. Conservation of stem-loop potential in introns of
snake venom phospholipase A2 genes. An application of FORS-D analysis. Mol. Biol. Evol.
12, 1157-1165.
Forsdyke, D. R. 1995c. Relative roles of primary sequence and (G+C)% in
determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of
different species. J. Mol. Evol.
41, 573-581.
Forsdyke, D. R. 1995d. Reciprocal relationship between stem-loop potential
and substitution density in retroviral quasispecies under positive Darwinian
selection. J. Mol. Evol. 41, 1022-1037.
Forsdyke, D. R. 1996a. Stem-loop potential in MHC genes: a new way of
evaluating positive Darwinian selection. Immunogenetics
43, 182-189.
Forsdyke, D. R. 1996b. Different biological species "broadcast"
their DNAs at different (C+G)% "wavelengths". J.
Theor. Biol. 178, 405-417.
Forsdyke, D. R. 1998. An alternative way of thinking about stem-loops in DNA.
A case study of the human G0S2 gene. J.
Theor. Biol. 192, 489-504.
Forsdyke, D. R. 2001a. Functional constraint and molecular evolution.
Encyclopedia of Life Sciences, Macmillan Reference Ltd., London.
Forsdyke, D. R. 2001b. The Origin of Species,
Revisited. McGill-Queen's
University Press, Montreal.
Forsdyke, D. R., Mortimer, J. R. 2000. Chargaff's legacy. Gene
261,
127-137.
Harris, H. 1994. An RNA heresy in the fifties. Trends. Biochem.
Sci. 19,
303-305.
Heximer, S. P., Cristillo, A. D., Russell, L., Forsdyke, D. R. 1996. Sequence
analysis and expression in cultured lymphocytes of the human FOSB gene (G0S3).
DNA Cell Biol. 15, 1025-1038.
Le, S. H. Maizel, J. V. (1989). A method for assessing the statistical
significance of RNA folding. J. Theor. Biol.
138, 495-510.
Liebovitch, L. S., Tao, Y., Todorov, A. T., Levine, L. 1996. Is there an
error-correcting code in the base sequence of DNA? Biophys. J.
71, 1539-1544.
Muller, H. J. 1922. Variation due to change in the individual gene. Am. Nat.
56, 32-50.
Naora, H., Deacon, N. J. 1982. Relationship between the total size of exons
and introns in protein-coding genes of higher eukaryotes. Proc. Natl. Acad.
Sci. USA 79,
6196-6200.
Pfeifer, K., Tilghman, S. M. 1994. Allele-specific gene expression in
mammals: the curious case of imprinted RNAs. Genes
Devel. 8, 1867-1874.
Salser, W. 1970. Discussion. Cold Spring Harb.
Symp. Quant Biol. 35, 19.
Santalucia, J., Allawi, H. T., Seneviratne, P. A. 1996. Improved nearest
neighbour parameters for predicting DNA duplex stability. Biochemistry
35, 3555-3562.
Seffens, W., Digby, D. 1999. mRNAs have greater negative folding free
energies than shuffled or codon choice randomized sequences. Nucleic Acids
Res. 27,
1578-1584.
Stoltzfus, A., Spencer, D. F., Zuker, M., Logsdon, J. M., Doolittle, W. F.
1994. Testing the exon theory of genes: the evidence from protein structure. Science
265,
202-207.
Weber, K., Kabsch, W. 1994. Intron positions in actin genes seem unrelated to
the secondary structure of protein. EMBO. J.
13, 1280-1286.
Winge, Ö. 1917. The chromosomes, their number and general importance. Compte.
Rend. Trav. Lab. Carlsberg 13, 131-275.
|