Adaptive Value of Polymorphism in Intracellular Self/Not-Self Discrimination?
D. R. FORSDYKE
Journal of Theoretical Biology (2001) 210,
425-434
(With copyright permission from Academic Press)
Based on an address given at the Second International Conference on Heat-Shock Proteins in the Immune Response, Farmington, CT, October 2000.
![]()
1. Within-Species Biological Discontinuity2. Differential Aggregation Hypothesis
3. Polymorphism and Individual-Specific Function
5. Antigenicity of Cancer Cells
6. Antigenicity of Viral Proteins
7. Resident Proteins as Intracellular "Antibodies"
A microbial pathogen species can adapt to its host species to the extent that members of the host species are uniform. Loss of this uniformity would make it difficult for a pathogen species to transfer, from one member of the host species to another, what it had "learned" through selection of its members with advantageous mutations. The existence of major histocompatibility complex (MHC) polymorphism indicates that non-uniformity within a species is an effective host defence strategy. By virtue of this molecular discontinuity among its members the host species can "present a moving target" to the pathogen. Many proteins other than MHC proteins show polymorphism, a phenomenon which has suggested that mutations in regions of protein molecules which do not affect overt function are neutral. However, in the context of the authors differential aggregation theory of intracellular self/not-self discrimination as previously applied to the problem of the antigenicity of cancer cells, such polymorphism should serve for the recruitment of subsets of self-antigens into the antigenic repertoire of an infected cell. These would act as "intracellular antibodies" by virtue of their weak, but specific, aggregation with pathogen proteins. Peptides from the self-antigens, as well as (or instead of) from the antigens of the pathogen, would then serve as targets for attack by cytotoxic T cells. Thus, polymorphism of intracellular proteins should be of adaptive value serving to amplify and individualise the immune response to intracellular pathogens. |
1. Within-Species Biological Discontinuity
|
Most microbial pathogens can
produce more progeny in a given time than their hosts. Having coevolved (Hafner
& Nadler, 1988), each pathogen species is adapted to its host species, and
should readily circumvent host defences through mutation and positive selection
of appropriately adapted members. Thus, the biological
continuity
(uniformity) of a host species places its members at risk in that microbial
adaptations acquired while infecting one member of the host species can assist
an attack on another member of that species. What the microbe "learns"
in one host, it can use to exploit the next. To this extent, biological
discontinuities
among members of the host species, which do not infringe species integrity,
should help them in their defence against microbial pathogens. One well-known biological
discontinuity is the phenomenon of major histocompatability complex (MHC)
polymorphism*.
Footnote:* A mutation is first
accepted in the DNA of a single individual and gives rise to an allele. If an
allele achieves an appreciable frequency in a population it constitutes a
polymorphism. A gene can be described as highly polymorphic if there are many
sets of such alleles in the population.
There being multiple alleles at a given MHC locus, most members
of a species are heterozygotes. The probability would be high that MHC-containing
cell membrane components acquired by a viral coat protein from one host, would
be seen as foreign ("not-self"), and so would alert the immune system
of a new host. This may have been elaborated over evolutionary time into the
sophisticated MHC-peptide system of existing species (Forsdyke, 1991; Obst et
al., 2000).
In the light of a previous
differential aggregation hypothesis of peptide selection for MHC loading (Forsdyke,
1995a), the present paper considers the possibility that much protein
polymorphism is not "neutral"
(Kimura, 1968; King & Jukes,
1969)
but, like MHC polymorphism, adapts the host organism as "a moving
target" to prevent pathogen
preadaptation. 2. Differential Aggregation Hypothesis
The hypothesis postulated that the concentration of each protein in the
crowded cytosol (Fulton, 1982) has been fine-tuned over evolutionary time to
approach the limits of its solubility, and yet avoid aggregating
"self" with "self." Various molecular chaperones, which
include the heat-shock proteins (HSPs), would quickly reverse any incipient
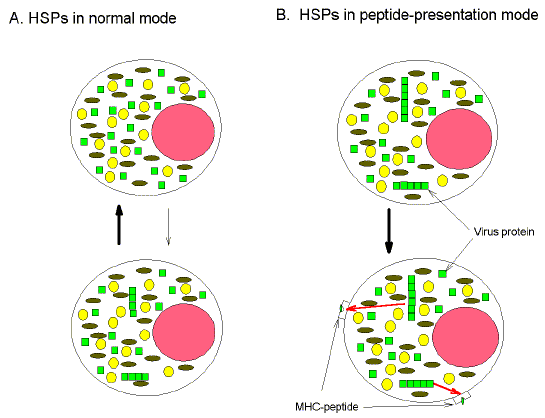
aggregation (Fig. 1a). (a)
"Self" proteins (small circles, ovals and squares) approach the limits
of their solubility in the crowded cytosol (area between the perimeters of the
large circle representing the cell wall and the medium-sized pink circle representing
the cell nucleus). HSPs (not shown) in "normal mode" act as molecular
chaperones to disassemble incipient aggregates. (b) "Not-self" viral
proteins (small squares) more readily cross the aggregation threshold than
"self" host proteins (small circles and ovals), and form larger
aggregates. These aggregates constitute self/not-self discriminatory signals,
resulting in a switch of HSPs (not shown) to peptide presentation mode, and the
triggering of various intracellular alarms, including the upregulation of MHC
class I protein expression. Aggregates are subject to proteosomal processing to
form peptides which, by way of HSP-peptide intermediates, are displayed at the
cell surface as MHC-peptide complexes.
On the other hand, virus proteins would tend to overstep
the aggregation threshold. This critical self/not-self discrimination event,
would trip various intracellular alarms resulting in pyrexia, proteosomal
degradation of aggregates to peptides, a switch of HSPs to peptide-binding mode,
up-regulation of MHC protein expression, and display of MHC-viral peptide
complexes at the cell surface (Fig. 1b). Thus, the cell would become a target
for attack by cytotoxic T cells, the host repertoire of which would include
cells able to recognize peptides from intracellular "self" proteins
(Forsdyke, 1995a; Sandberg et al.,
2000).
The HSP mode switch could require either a change from the synthesis of
constitutive HSPs to the synthesis of inducible HSPs (Menoret et al.,
1995), or a change in properties of HSP molecules
(Spiess et al.,
1999).
To prevent inadvertent aggregation of self- proteins following the tripping of
the alarm, their synthesis would be down-regulated (e.g. the corresponding genes
might have accepted mutations inhibiting the binding of RNA polymerase under
alarm conditions); this would decrease the concentrations of self-proteins (a
phenomenon perhaps of consequence for understanding the evolution of genetic
dominance; Forsdyke, 1994a; Hurst & Randerson, 2000). Similarly, viruses
would tend to accept mutations that decrease the chance of their proteins
overstepping the aggregation threshold. This problem, not addressed in the
initial hypothesis (Forsdyke,
1995a,b), will be considered in Section 8. 3. Polymorphism and Individual-Specific Function
For simplicity biological species are here considered as composed of nucleic
acids and proteins, and other molecules are disregarded. Members of a biological
species have many similar anatomical and physiological features and,
accordingly, they have similar proteins, which are responsible for this
phenotype. Some of these proteins are also found in members of other biological
species, where they have similar functions. Often the name of a protein
indicates its overt function. Thus the enzyme lactate dehydrogenase oxidises
lactate to pyruvate, a function required in many taxa. The performance of this
function places the enzyme under strict stereochemical constraint. Especially
within a species, but often also between species, there is high evolutionary
conservation of amino acids in certain regions of the molecule (e.g. the
catalytic centre and regions which support the integrity of the catalytic centre;
Fersht, 1998). Organisms with mutations in these regions are usually negatively
selected ("purifying selection") in the course of evolution. Apart from their overt specific functions, most protein molecules have other
specific functions. For example, if it is advantageous for a protein to be
localized within its cell, or to form dimers, then amino acids involved in these
specific intermolecular reactions will be evolutionarily conserved. Thus, the protein molecules of a biological species have regions that are
conserved among species members, and regions that are less conserved among
species members
(i.e. polymorphism; Harris, 1966; Lewontin & Hubby, 1966; Forsdyke,
2001). Some less conserved regions could play a non-specific structural role, so
that amino acids in these regions would act more by virtue of being neutral
"placeholders" than by virtue of their specific chemical
characteristics.
Alternatively, the less conserved regions could have no overt
role, either specific or non-specific, in the regular day-to-day life of the
organism (i.e. they would appear completely neutral in that their deletion would
not affect overt function). Such neutral and completely neutral regions tend to
be solvent accessible and surface located (Goldman et al., 1998;
Bustamente et al., 2000; Alvarez-Valin et al., 2000). This
indicates a potential to engage in intermolecular interactions. The greater the
range of the polymorphism, the greater the range of this potential among members
of a biological species.
Indeed, the assumption of neutrality may not be valid. Polymorphism involving
less conserved regions may have a specific function. Polymorphism
within
cells might serve the organism in much the same way that MHC polymorphism at the
cell surface serves the organism. The fact of non-conservation implies that,
like MHC polymorphism, the specificity is likely to be
individual-specific,
and is not shared by many other members of the species. Although this
polymorphism might at one time have been neutral, it is argued here that such
territory would soon have been trespassed upon to serve adaptive needs.
The biological effect of a molecule usually depends on its functional
potential and its concentration. By accepting appropriate mutations the
corresponding gene can "tune" or "fine-tune" either or both
of these properties, and hence modify the biological effect (change the
phenotype). For present purposes, changes in functional potential will be
emphasized. Consider a biological species in which protein molecule
A would
increase the genetic fitness of species members if it interacted in the
cytoplasm with protein molecule B. For example, localization of one enzyme in a
metabolic pathway close to another enzyme (for which its product provides a
substrate) might improve the efficiency of the pathway (Scott & Forsdyke,
1978; Kisters-Woike et al., 2000). Let there be a germline mutation
resulting in a change (A
to
A'), such that a complex
A'B can now be formed. Having arranged the structure of molecule
A such that
it can now react with molecule B, the "hand of evolution" has achieved
a significant advance. Such evolutionary "tuning" should occur
quite rapidly because of the immediate selective advantage it would confer.
Thus, over time species members with A' would be positively selected and would
replaced species members with A. A'
+ C = A'C
+ Weak A'
+ D
= A'D
+ Weak A'
+ E
= A'E
+ Weak A'
+ F
= A'
+ G
= .
. .
. .
. A'
+ Z
=
However, the crowded cytoplasm contains many thousands of protein types (e.g.
A-Z; Fig. 2). It is probable that a small subset of proteins (C, D, E) with no
functional interest in A, would, by virtue of the primary change in A,
by
chance, now become able to interact weakly with it, to form complexes
A'C,
A'D and A'E. These unwanted reactions, albeit weak, might unduly immobilize
the interacting molecules, to the marginal detriment of their primary overt
functions. The advantage of forming A'B complexes would so far outweigh the
disadvantages of forming A'C, A'D and A'E complexes, that the differential
survival of individuals with A' would not be threatened. Nevertheless, it
would be expect that, concomitant with and following the fixation of the gene
encoding A' in the species, there would be an on-going random process of
mutational counter-adaptation by genes encoding C, D and E to decrease their
affinity for A', provided this could be achieved without compromising their
primary functions (i.e. the mutations would affect "neutral,"
potentially polymorphic, residues).
Such random "fine-tuning"
(C
to
C',
D
to
D', E
to
E')
would be likely to occur in
different
individuals at
different
times, and would confer virtually insignificant adaptive advantages on those
individuals. Thus individuals with constitutions A'B'CD, A'BC'D,
A'BCD'
would be favoured only slightly over the general population of individuals of
constitution A'BCD (i.e. the adaptations would initially be "near
neutral"; Ohta, 1995). This biological discontinuity (polymorphism) would
appear relatively stable. Over many generations, however, genetic recombination
would create, by chance, alliances of these fine-tuned proteins so that
individuals of constitution A'B'C'D' would eventually emerge, and would
be marginally advantaged. The proportion of their offspring would increase in
the population, reducing the extent of the biological discontinuity. While this slow
proximate
fine-tuning was in progress, a seemingly
unending chain of further fine-tunings would be occurring. In the same way that
the initial change A
to A' provoked
counter-adaptations in C, D and E, so the changed forms C',
D' and E',
should
each
provoke corresponding counter-adaptations in
individuals
containing them. These molecules
(F-N) whose functions would themselves now be marginally
compromised, would in-turn need to counteradapt to F'-N'. This, in turn ,
would provoke further counter-adaptations, etc., etc..
Thus, at any time-point there would be an intracellular network of proteins
the potentially polymorphic regions of which the "hand of evolution"
would be fine-tuning in order that the proteins not react, however weakly, with
others. New positive mutations of type A
to A' would
from time-to-time set off new chains of adaptations and counter-adaptations.
However, if the environment remained relatively constant the organism would
asymptotically approach a state of perfect adaptation. Then the frequency of the
positive mutations would decrease, and the initial round of proximate
counter-adaptations (C', D' and E') would approach completion. Individuals
of type A'B'C'D' would come to dominate the species which would loose
this particular biological discontinuity.
In this way many natural species would
be fine-tuned and appear well adapted to their environments. Some plants in this
state we might designate as "weeds." They grow rapidly and form seed
even in nutritionally unfavourable conditions. On the other hand, domestic
plants, relative late-comers on the evolutionary scene, usually grow more
slowly, even in nutritionally favourable conditions.
Could this intrinsic tendency towards polymorphism of certain surface-located
amino acid residues, which may engage in weak, random, and apparently
non-functional, interactions, have been turned to the further advantage of the
organism? To approach this question, we first reconsider previous work (Forsdyke,
1999) to show how, at the level of the individual, polymorphism might
act to increase the effective antigenicity of mutations in oncogenes (Section
5). It will then be shown that the same principle might apply in the enhancement
of the effective antigenicity of virus proteins. The designation by the host of
a virus protein as "not-self," may involve both quantitative factors
(i.e. its concentration exceeding a solubility threshold, which the virus might
avoid by appropriate mutation), and qualitative factors (the recruitment of
various polymorphic host proteins into protein aggregates, which the virus might
not avoid by mutation; see Sections 6-8). 5. Antigenicity of Cancer Cells
Consider normal cells that have become potentially cancerous because of a
mutation in a single oncogene. In practice, a number of such mutational switches
involving different oncogenes might need to be "thrown" for cells to
manifest the full cancer phenotype (Klein & Klein,
1985). Since oncogenes
are a distinct subset of genes common to all members of a species, it is
attractive to suppose that the primary mutation in an oncogene (A
to
A') would be recognized by an intracellular self/not-self discrimination
system (involving differential aggregation of A';
Forsdyke, 1994b,
1995a,b) so
that a peptide from A would be displayed in association with MHC proteins at
the surface of the cancer cell. This MHC-peptide complex would be recognized by
cytotoxic T-cells, which would then multiply and destroy the potential cancer
cell or its progeny before they could form a tumour. This predicts
cancer-specific antigens.
However, the cancer antigens recognized by cytotoxic T cells tend to be
neither
cancer-specific,
nor
cancer type-specific. Rather they are specific to
the
individual
with cancer. Antigens specific for cancers in general, or
for particular cancer types, are hard to find and do not appear to serve as
useful primary targets for immune attack . This suggests antigen polymorphism since, if (say)
B were present as
B (rather than as B') in an individual whose cancer was being attacked with
respect to antigens E, F and G, then it is hard to see how
B would be excluded from the attack.
Yet, when cancer antigens were purified, many were found to correspond to
heat-shock proteins, which are not polymorphic. Remarkably, the heat-shock
proteins could confer individual-specific protective immunity against the cancer
of origin. Srivastava et al., (1998) noted: "The observed specificity of immunogenicity of cancer-derived HSPs
would suggest that they display somatic polymorphism such that HSPs would
differ between cancers and normal tissues and from one cancer to another.
However, extensive sequencing studies of HSP cDNAs of cancers and normal
tissues did not support this idea. What is the origin of the specificity of
immunogenicity?"
The specificity of immunogenicity was found to derive from the association
with the HSPs of peptide fragments of the products of non-oncogenes. In view of
the widespread evolutionary conservation of HSP genes, it was suggested that
HSPs might have constituted a prototypic system for the processing of peptides,
which evolved into the contemporary MHC-peptide system. The modern role of HSPs
might be to transfer peptides to MHC molecules, either within the cell of
peptide origin for presentation by that cell to cytotoxic T cells, which would
destroy the cell and proliferate (Vanbuskirk et al.,
1989), or within
other cells (dendritic cells) for presentation by those cells (Binder et al.,
2000; Singh-Jasuja et al., 2000). By transferring the peptide signal
beyond the cell of origin to a "professional" antigen-presenting cell,
HSPs would serve to amplify the signal, causing specific cytotoxic T cells to
proliferate further.
To explain the apparently random recruitment of
non-oncogenes into the
antigenic repertoire of the cancer cell, Srivastava proposed that the cancer
phenotype predisposes to mutation (Lengauer et al., 1998; Tomlinson & Bodmer,
1999) genes which are unlikely to be related functionally to the genes whose
primary mutation results in cancer. These non-oncogenes, by virtue of the
mutations they contained, would not affect oncogenicity per se, but would
mark cancer cells as foreign for recognition by an immune system considered to
have been educated to distinguish "not-self", from "self"
(Srivastava,
1993, 1996). However, it was argued
(Forsdyke, 1999), that this formulation is
incorrect both on theoretical grounds and on experimental grounds, namely,
that the individual-specific non-oncogene antigens derived from cancer cells
with a mutation in an oncogene are usually not mutated (Srivastava, 1996;
Srivastava et al., 1998; Zeh et al., 1999). The requirement that genes must mutate to "not-self" in order to be
able to trigger immune defences would not be necessary if Thus,
instead of describing potential individual-specific antigenic phenotypes as A'B'C'D', A'E'F'G', A'H'I'K', we would write
A'BCD, A'EFG, A'HIK. This amends an earlier formulation (Forsdyke,
1999) where differences between
individuals in the original mutation (A
to
A',
A
to
A'',
A
to
A''') were invoked to produce
antigenic phenotypes A'BCD, A''EFG, A'''HIK (i.e. the mutational
change conferring oncogenicity could be either A', A'', or A''', and
these different forms of A would recruit different sets of proteins for
targeting by cytotoxic T cells).
Following the argument of Section 4, the
same
mutation
A' in
different individuals should suffice to recruit sets of polymorphic proteins
(from among B-K) into the respective antigenic phenotypes. The
individual-specific nature of the antigenicity would appear readily explicable
in terms of the natural polymorphic tendency of proteins with respect to amino
acid residues which are not under strict functional constraint as related either
to their generally-recognized named role (e.g. lactic dehydrogenase), or to
their ability to engage in specific intermolecular reactions related to this
function. It should be noted that the initial hypotheses of Srivastava (1993)
and of Forsdyke (1999), both postulate an apparent polymorphism generated in
somatic time in response to a mutated oncogene, whereas the present version
postulates a real preexisting polymorphism generated over evolutionary time. 6. Antigenicity of Viral Proteins
HSPs may be involved in the presentation, not only of
tumour-derived
antigens, but also of antigens from virally-infected cells (Ciupitu et al.,
1998). A protective role of polymorphic intracellular proteins should be very
important in virus infections. A viral protein (A) can be considered in the same
way as a mutant oncogene, so that host proteins B, C and D would be recruited
into the antigenic repertoire of the infected cell. A mutation in a viral
protein A to A' which might
prevent
it interacting with host proteins
B, C and D, might be of little value when the virus infected its next host. In
this host these proteins might exist as polymorphic forms B', C' and
D',
which might not interact with A. Thus, in this host the polymorphic proteins
recruited into the antigenic repertoire might be E, F and G. In another host
H,
I and J might be recruited.
In contrast, the
extracellular
recognition of a not-self viral protein
("antigen") involves a normal resident protein ("antibody"),
which interacts with a discrete part ("epitope") of the intact
antigen. In this case, the antibody protein (immunoglobulin) has evolved for
this specific purpose and, as far as we are aware, has no other primary purpose.
Its polymorphism has been developed to the extreme by the evolution of variable
region genes with the potential to diversify further during somatic time (Tonegawa,
1988).
In the case of
intracellular
proteins there is no known dedicated
molecular species of an antibody nature. So proteins with normal
functions in the economy of the cells would be co-opted for the aggregation
function. Host proteins B, C and D can be regarded as intracellular
"antibodies" of very low specificity, which happen to have sufficient
complementarity with A to form a complex in the crowded cytosol (an environment
highly conducive to intermolecular interactions;
Forsdyke,
1995a).
Just as extracellular antigen-antibody complexes, having labelled intruders
as "not-self," trigger amplifying inflammatory responses, so
multimolecular intracellular aggregates (A'B, A'C or A'D) should suffice
to initiate intracellular "inflammation." This would lead to MHC
presentation of their peptides (Townsend et al.,
1990), and the release
of chemokines such as G0S19/MIP1" (Cook et
al., 1995; Heximer et al., 1998). To the extent that intracellular
"antibodies," MHC proteins and chemokines are
not
polymorphic,
successful viral counter-adaptations would be expected (Davis-Poynter &
Farrell, 1996), and there would exist the necessary elements for an arms race
between pathogen and prey, leading to positive Darwinian selection of variants
of both (Forsdyke, 1995c,
1996). To the extent that the proteins
are
polymorphic, viral counter-adaptations would less likely be successful.
It should be noted that polymorphic MHC proteins usually display differential
peptide binding (Townsend et al.,
1990), with the result that one
individual may display a peptide which another (with a different MHC haplotype)
cannot. It is not envisioned here that this cause of the differential
presentation of peptides from self-antigens plays as important a role as the
intracellular "antibodies." Consistent with this, species with a
limited number of MHC genes can remain healthy (Kaufman,
2000). 7. Resident Proteins as Intracellular "Antibodies"
Solubility, a fundamental protein property, is at the centre of the
differential aggregation hypothesis of self/not-self recognition (Forsdyke,
1995a, b). As their limit of solubility is approached, molecules of resident
cellular protein types would tend to aggregate. The aggregations would be
protein-specific, in that proteins aggregate
like-with-like,
leaving other protein types unaggregated (Lauffer, 1975; Leikin &
Parsegian, 1994). The growing aggregates would tend to become insoluble forming
precipitates or cellular "inclusion
bodies." This phenomenon has long
been exploited by biochemists to purify different protein types from mixtures by
differential precipitation (e.g. with polyethylene glycol). The specificity
appears similar to that required for the formation of pure crystals from
molecular mixtures, a process which seems to require structural regularities,
and perhaps molecule-specific vibrations, or resonances (Israelachvili &
Wennerstrm, 1996).
When the sequences of two alleles randomly chosen from the human population
are compared, there are approximately two variations per kb. Thus, in a large
population, many 1 kb sequences would be expected to differ from the canonical
sequence (Rowen et al., 1997; Sunyeav et al.,
2000). Polymorphism
is widespread, but is the extent of polymorphism sufficient for the role
considered here? For simplicity we have considered only one viral gene (A), and
the recruitment of three host proteins (e.g. B, C and D). However, viruses
generally synthesize many different proteins at different stages of their life
cycle, each with the potential to recruit a distinct subset of host proteins.
Furthermore, the recruitment of only one host protein should suffice to trigger
an antiviral response in that host, while, because of polymorphism, the same
protein might not be recruited in a subsequent host (which might present
peptides from another polymorphic protein).
Thus, although a virus might accept
mutations which change its proteins
quantitatively
(i.e. so that they do
not exceed a concentration threshold beyond which self-aggregation would occur),
its acceptance of mutations which change its proteins
qualitatively
(i.e.
change its protein sequences to decrease their ability to recruit host proteins
into aggregates) might confer no adaptive advantage if such host proteins were
polymorphic. When confronted with such polymorphism, to evade detection by the
host more profound decreases in viral protein quantities would be required. Unlike viruses, an oncogene activated by mutation is unlikely to mutate
further, so confusing the defences of the host cell. Indeed, it would be
beneficial for one or more self-proteins, to the extent that their primary
functions were not impaired, to accept mutations which promote their
interactions with (and hence potential coaggregation with) such mutated oncogene
proteins. If primary oncogene mutations have in the past diminished reproductive
success, this inherited adaptation of non-oncogenic self-proteins would tend to
make cancer a disease of post-reproductive life.
Since such adaptation of
self-proteins could confer an advantage, the corresponding alleles would be
positively selected and monomorphism should be developed more than expected
under a strictly neutral mutation hypothesis. Modelling studies tend to support
this view, but it is noted that "what the selection might be remains
unclear" (Eyre-Walker, 1999;
Akashi, 1999). Currently, most models do not
distinguish between the many potential, sometimes competing, non-neutral
pressures on genomes, which include GC-pressure, purine-loading pressure,
fold-pressure, and the "polymorphic pressure" considered here
(Forsdyke & Mortimer,
2000). 10. Another Self/Not-Self Discrimination Signal? A
virus appears in a cell both as nucleic acid and as protein. While beyond
the scope of the present discussion, it should be noted that the formation of
double-stranded RNAs between a virus RNA and host RNAs (for refs. see Forsdyke
& Mortimer, 2000; Lao & Forsdyke, 2000; Cristillo et al., 2001),
provides another potential self/not-self discriminatory signal which might
supplement or synergize with the postulated differential protein aggregation
signal (Forsdyke, 2000; (Click Here) and see
End
Note (below)). REFERENCES AKASHI, H. (1999). Within- and between-species DNA sequence variation and the
footprint of natural selection. Gene 238, 39-51. ALVAREZ-VALIN, F., TORT, J. F. & BERNARDI, G. (2000). Nonrandom spatial
distribution of synonymous substitutions in the GP63 gene from Leishmania. Genetics
155, 1683-1692. BINDER, R. J., HAN, D. K. & SRIVASTAVA, P. K. (2000). CD91: a receptor
for heat shock protein gp96. Nature Immun. 1, 151-155. BUSTAMENTE, C. D., TOWNSEND, J. P. & HARTL, D. L. (2000). Solvent
accessibility and purifying selection within proteins of Escherichia coli
and Salmonella enterica.
Mol. Biol. Evol. 17, 301-308. CIUPITU, A. M., PETERSSON, M., ODONNELL, C. L., WILLIAMS, D. K.,
JINDAL, S., KIESSLING, R. & WELSH, R. M. (1998). Immunization with a lymphocytic
choriomeningitis virus peptide mixed with heat shock protein 70 results in a
protective antiviral immunity and specific cytotoxic lymphocytes.
J. Exp. Med. 187,
685-691. COOK. D. N., BECK, M. A., COFFMAN, T. M., KIRBY, S. L., SHERIDAN, J. F.,
PRAGNELL, I. B. & SMITHIES, O. (1995). Requirement of MIP1"
for an inflammatory response to viral infection. Science 269, 1583-1585.
CRISTILLO, A. D., MORTIMER, J. R., BARRETTE, I. H.,
LILLICRAP, T. P. &
FORSDYKE, D. R. (2001). Double-stranded RNA as a not-self alarm signal: to
evade, most viruses purine-load their RNAs, but some (HTLV-1,
EBV) pyrimidine-load. J.
theor. Biol. 208, 475-491. DAVIS-POYNTER, N. J. & FARRELL, H. E. (1996). Masters of deception: a
review of herpesvirus immune evasion strategies. Immun. Cell Biol. 74,
513-522.
EYRE-WALKER, A. (1999). Evidence of selection on silent site base composition in
mammals: potential implications for the evolution of isochores and junk DNA.
Genetics 152, 675-683. FERSHT, A. (1998). Structure and Mechanism in Protein
Science. New
York: W. H. Freeman. FORSDYKE, D. R. (1991). Early evolution of MHC polymorphism.
J. theor. Biol.
150, 451-456.
FORSDYKE, D. R. (1994a). The heat-shock response and the molecular basis of
genetic dominance. J.
theor. Biol. 167, 1-5. FORSDYKE, D. R. (1994b). Relationship of X chromosome dosage compensation to
intracellular self/not-self discrimination: a resolution of Mullers paradox? J.
theor. Biol. 167, 7-12.
FORSDYKE, D. R. (1995a). Entropy-driven protein self-aggregation as the basis
for self/not-self discrimination in the crowded cytosol. J. Biol. Sys.
3, 273-287.
FORSDYKE, D. R. (1995b). Fine tuning of intracellular protein concentrations, a collective protein function involved in aneuploid lethality, sex-determination
and speciation? J. theor. Biol. 172, 335-345.
FORSDYKE, D. R. (1995c). Conservation of stem-loop potential in introns of snake
venom phospholipase A2 genes: an application of
FORS-D analysis. Mol.
Biol. Evol. 12, 1157-1165. FORSDYKE, D. R. (1996). Stem-loop potential in MHC genes: a new way of
evaluating positive Darwinian selection. Immunogenetics 43, 182-189.
FORSDYKE, D. R. (1999). Heat shock proteins as mediators of aggregation-induced danger signals: implications of the slow evolutionary fine-tuning of
sequences for the antigenicity of cancer cells. Cell Stress Chaperones 4, 205-210. FORSDYKE, D. R. (2000). Double-stranded RNA and/or heat-shock as initiators
of chaperone mode switches in diseases associated with protein aggregation.
Cell
Stress Chaperones 5, 375-376. FORSDYKE, D. R. (2001). Functional constraint and molecular evolution.
Encyclopedia of Life Sciences. Macmillan Reference Ltd., London.
FORSDYKE, D. R. & MORTIMER, J. R. (2000). Chargaffs legacy. Gene 261,
127-137. FULTON, A. (1982) How crowded is the cytoplasm?
Cell 30,
345-347.
GOLDMAN, N., THORNE, J. L. & JONES, D. T. (1998). Accessing the impact of
secondary structure and solvent accessibility on protein evolution.
Genetics
149, 445- 458. HAFNER, M. S., & NADLER, S. A. (1988). Phylogenetic trees support the
coevolution of parasites and their hosts. Nature 332, 258-259. HARRIS, H. (1966). Enzyme polymorphisms in man.
Proc. R. Soc. Lond. B.
164, 298-310.
HEXIMER, S. P., ERNST, B. D., RUSSELL, L. & FORSDYKE, D. R. (1998). The
normal copy of the G0S19-3-associated, CpG island-containing, upstream
sequence is downstream of G0S19-2/MIP1a in association
with a TRE17 oncogene. DNA Cell
HURST, L. D. & RANDERSON, J. P. (2000). Dosage, deletions and dominance:
simple models of the evolution of gene expression.
J. theor. Biol. 205,
641-647. ISRAELACHVILI, J. & WENNERSTR_
M, H. (1996). Role of hydration and water structure in biological and colloidal interactions. Nature 379,
219-224.
KAUFMAN, J. (2000). The simple chicken major histocompatibility complex: life
and death in the face of pathogens and vaccines.
Phil. Trans. R. Soc. Lond. B.
355, 1077-1084. KIMURA, M. (1968). Evolutionary rate at the molecular level.
Nature
217,
624-626.
KING, J. L. & JUKES, T. H. (1969). Non-Darwinian evolution. Science 164,
788-798.
KISTERS-WOIKE, B., VANGIERDEGOM, C. & MULLER-HILL, B. (2000). On the
conservation of protein sequences in evolution. Trends.
Biochem. Sci. 25,
419-421. KLEIN, G. & KLEIN, E. (1985). Evolution of tumours and the impact of
molecular oncology. Nature 315, 190-195. LAO, P. J. & FORSDYKE, D. R. (2000). Thermophilic bacteria strictly obey
Szybalskis transcription direction rule and politely purine-load RNAs with both adenine
and guanine. Genome Res. 10, 228-236. LAUFFER. M. A. (1975). Entropy-Driven Processes in Biology. New York:
Springer-Verlag.
LEIKIN, S. & PARSEGIAN, V. A. (1994). Temperature-induced complementarity as
a mechanism for biomolecular assembly. Proteins 19, 73-76.
LENGAUER, C., KINZLER, K. W. & VOGELSTEIN, B. (1998). Genetic instabilities
in human cancers. Nature
396, 643-649. LEWONTIN, R. C. & HUBBY, J. L. (1966). A molecular approach to the study
of genic heterozygosity in natural populations. II. Amount of variation and degree of
heterozygosity in natural populations of Drosophila pseudoobscura. Genetics
54, 595- MENORET, A., PATRY, Y., BURG, C. & LE
PENDU, J. (1995). Co-segregation of tumor immunogenicity with expression of inducible but not constitutive hsp70
in rat colon carcinomas. J. Immun. 155, 740-747. NEWNAM, G. P., WEGRZYN, R. D., LINDQUIST, S. L. &
CHERNOFF, Y. O. (1999). Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion
curing. Mol. Cell. Biol. 19, 1325-1333. OBST, R., NETUSCHIL, N., KLOPFER, K., STEVANOVI,
S. & RAMMENSEE, H-G. (2000). The role of peptides in T cell alloreactivity is determined by
self-major histocompatability complex molecules. J. Exp. Med. 191, 805-812.
OHTA, T. (1995). Synonymous and nonsynonymous substitutions in mammalian genes
and the nearly neutral theory. J. Mol. Evol. 40, 56-63.
ROWEN, L., MAHAIRAS, G. & HOOD, L. (1997). Sequencing the human genome.
Science
278, 605-607. SANDBERG, J. K., FRANKSSON, L., SUNDBACK, J.,
MICHAELSSON, J., PETERSSON, M., ACHOUR, A.,
WALLIN, R. P. A., SHERMAN, N.E., BERGMAN, T.,
JORNVALL, H., HUNT, D. F., KIESSLING, R. & KARRE, K. (2000). T cell tolerance based on avidity thresholds rather than complete
deletion allows maintenance of maximal repertoire diversity.
J. Immun. 165,
25-33. SANTOSO, A., CHIEN, P., OSHEROVICH, L.Z. &
WEISSMAN, J. S. (2000). Molecular basis of a yeast prion species barrier. Cell
100,
277-288.
SCOTT, F. W. & FORSDYKE, D. R. (1978). The rate of deoxyribonucleic acid
synthesis by cultured Chinese Hamster ovary cells. Biochem. J. 170, 545-549. SINGH-JASUJA, H., SCHERER, H.U., HILF, N.,
ARNOLD-SCHILD, D., RAMMENSEE, H.-G., TOES,
R.E.M. & SCHILD, H. (2000). The heat shock
protein gp96 induces maturation of dendritic cells and down-regulation of the gp96
receptor. Eur.J. Immunol.
30, 2211-2215. SPIESS, C., BELL, A. & EHRMANN, M. (1999). A temperature-dependent switch
from chaperone to protease in a widely conserved heat-shock protein.
Cell 97,
339-347. SRIVASTAVA, P. K. (1993). Peptide-binding heat shock proteins in the
endoplasmic reticulum: role in immune response to cancer and in antigen presentation. Adv.
Cancer Res. 62, 153-177.
SRIVASTAVA, P. K. (1996). Do human cancers express shared protective antigens?
Or the necessity of remembrance of things past. Sem. Immunol. 8,
295-302.
SRIVASTAVA, P. K., MENORET, A., BASU, S., BINDER, R. T. &
MCQUADE, K. L. (1998). Heat shock proteins come of age: primitive functions acquire new roles
in an adaptive world. Immunity 8, 657-665.
SUNYEAV, S. R., LATHE, W. C., RAMENSKY, V. E. &
BORK, P. (2000). SNP frequencies in human genes: an excess of rare alleles and differing modes of
selection. Trends. Genet. 16, 335-337. TOMLINSON, I. & BODMER, W. (1999). Selection, the mutation rate and
cancer: ensuring that the tail does not wag the dog. Nature Med. 5, 11-12. TONEGAWA, S. (1988). Somatic generation of immune diversity.
Biosci.Rep.
8, 3-26. TOWNSEND, A., ELLIOTT, T., CERUNDULO, V., FOSTER, L., BARBER, B. &
TSE, A. (1990). Assembly of MHC class I molecules analyzed in vitro. Cell
62, 285-295.
WARRICK, J. M., CHAN, H.Y.E., GRAY-BOARD, G. L.,
CHAI, Y., PAULSON, H.L. & BONINI, N. (1999). Suppression of polyglutamine-mediated neurodegeneration
in Drosophila by the molecular chaperone HSP70. Nature Genetics
23, 425-428. VANBUSKIRK, A., CRUMP, B. L., MARGOLIASH, E. & PIERCE, S. K. (1989). A
peptide-binding protein having a role of antigen presentation is a member of
the HSP70 heat shock family. J. Exp. Med. 170, 1799-1809. ZEH. H. J., PERRY-LALLEY, D., DUDLEY, M. E., ROSENBERG, S. & YANG, J. C.
(1999). High avidity CTLs for two self-antigens demonstrate superior in vitro
and in vivo antitumor efficiency. J. Immunol. 162, 989-994.
End Note
2001:
dsRNA in Huntington's
Disease
Evidence
consistent with self/not-self discrimination acting at
both
protein and RNA levels was provided by Peel et al. (2001)
Nucleic
Acids Res.
10,
1531-1538. "Double-stranded RNA-dependent protein kinase, PKR, binds
preferentially to Huntington's disease (HD) transcripts and is activated in HD
tissue."
End Note
2008:
Conservation of Degree of
Macromolecular Crowding
The
fine-tuning of the concentration of a cytosolic protein to that of its companion
proteins, predicts high conservation of the degree of macromolecular crowding.
This has been found in the case of cells from different mammalian species (see
Guigas, Kalla & Weiss (2007)
FEBS
Letters
581,
5094-5098. "The degree of macromolecular crowding in the cytoplasm and
nucleoplasm of mammalian cells is conserved."
End Note
2009:
Interaction Promiscuity The above
"subset of proteins (C, D, E) with no
functional interest in A"can be said to engage in "pairwise"
interactions with A. They do not mutually interact as would be the case if they
formed a functional network. This was supported by a study of factors affecting
"dosage sensitivity" of gene products (Vavouri et al. 2009). However,
the propensity to self-aggregate did not score highly, perhaps reflecting the
inapplicability to crowded cytoplasmic conditions of the "TANGO"
algorithm that was employed. Marcotte, E. M. &
Tsechansky, M. (2009) Disorder, promiscuity and toxic partnerships. Cell
138, 16-18. Vavouri, T., Semple, J. I.,
Garcia-Verdugo, R. & Hehner, B. (2009) Intrinsic protein disorder and
interaction promiscuity are widely associated with dosage sensitivity. Cell
138, 198-208.
End Note
2012:
Need for Avoidance
of Interactions Dictates More Conservation of Highly Expressed Proteins
Another
prediction of the hypothesis is that proteins contributing most to macromolecular crowding
will be most conserved.
This means that highly expressed proteins are likely to be more conserved than
lowly expressed proteins. This has been found to apply, at least for unicellular
organisms (see Yang, Liao, Zhuang & Zhang (2012)
Proc. Natl Acad Sci USA
"Protein misinteraction avoidance causes highly expressed proteins to evolve
slowly"). Citing numerous studies, it is stated that "Unexpectedly, the
strongest determinant of the rate of protein sequence evolution was found to be
its expression level" so that there is a "negative correlation between the
expression level of a protein and its evolutionary rate (E-R anticorrelation)."
End
Note (July 2016)
Neoantigens Sometimes Similar Next: Immunity as a Function of the
Unicellular State (2002) (Click Here) Return to: Theoretical Immunology Index (Click Here) Return to: Homepage (Click Here) Last edited on
17 Jan 2018
by D. R. Forsdyke |