The key to the origin of species is the phenomenon of hybrid sterility.
A mule is the hybrid formed by crossing a healthy fertile horse and a
healthy fertile ass. The mule is sterile showing that, despite their
health, the parents are reproductively isolated
from each other (but not necessarily from other members of their respective species). A modified chromosomal theory requiring diffuse differences only in single bases was presented in the Journal of Theoretical Biology (1996). This postulated that (C+G)% is the "accent" of DNA, which, like the accent of human beings (metaphorically speaking), can affect reproductive success (see Eliza Doolittle, below). An important path to this theory was to follow the approach of the molecular biologists in the 1940s and 1950s. They studied the simplest possible biological forms - the viruses that infect bacteria. So, regarding the speciation question, we sought evidence on this from viruses - in this case viruses that infected eukaryotic cells. In chemical terms, (C+G)% differences have a profound affect on the ability of a duplex DNA molecule to extrude the stem-loop structures by which homologous chromosomes first recognize each other at meiosis. Donald Forsdyke |
 Theories of
this phenomenon are either "genic" or "chromosomal".
Until 1996, chromosomal theories had required differences in large segments (e.g.
deletions, translocations), which might sometimes be seen on examination with a standard
light microscope.
Theories of
this phenomenon are either "genic" or "chromosomal".
Until 1996, chromosomal theories had required differences in large segments (e.g.
deletions, translocations), which might sometimes be seen on examination with a standard
light microscope. Different Biological Species "Broadcast" their DNAs at Different (C+G)% "Wavelengths"
by D. R. FORSDYKE
J. Theor. Biol. (1996) 178, 405-417. (With copyright permission from Academic Press.)
2. PRIMARY AND SECONDARY COMPONENTS OF INFORMATION
3. CODON CHOICE AND THE NEUTRALIST SELECTIONIST DEBATE.
4. SEX AND RECOMBINATION REPAIR
6. EVIDENCE FOR STEM-LOOP "KISSING" MODEL
7. HOW (C+G)% DIFFERENCES MIGHT IMPEDE RECOMBINATION.
8. THE EARLY EVOLUTION OF INTRONS
10. MAJOR PRESSURES ON DNA ARE SELECTIVE
Radio can be used as a metaphor for the transmission of information by DNA through time and space. Just as different radio transmitters broadcast at different wavelengths to prevent interference, so different biological species "broadcast" their DNAs at different (G+C)% "wavelengths" to prevent recombination. It is postulated that species differences in (G+C)% prevent recombination. First, evidence is presented supporting the early Crick-Sobell stem-loop model for genetic recombination, which proposes that the rate-limiting step in recombination is the recognition ("kissing") of complementary sequences in the loops of stem-loop structures extruded from supercoiled DNA. Then, various ways in which differences in (G+C)% might impede complementary loop interactions are outlined. The strength of the postulate is that it brings together a variety of disparate observations in fields that have not previously been seen as related. Thus explanations are apparent for why most mutations are not selectively neutral (the "neutralist/selectionist" debate), why introns were present in the earliest genes (the "introns-early / introns-late" debate), and the origin of species. |
- What is the molecular basis of speciation?
- What is the molecular basis of recombination?
- Are most accepted mutations in DNA selectively neutral?
- Were introns present in the earliest genes?
-
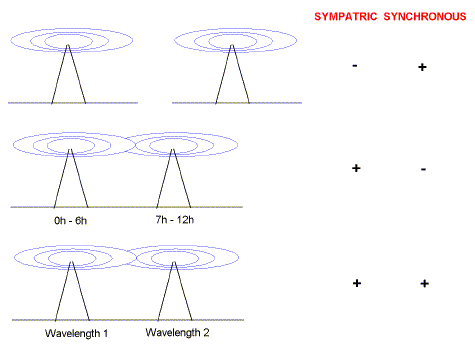
The second transmitter could operate at the same time as (synchronously with) the first
transmitter, but in a distant geographic area ("allopatrically")
beyond the range of the first transmitter
- The first and second transmitters could transmit in the same geographical area ("sympatrically"), but would agree to transmit at different
times (asynchronously).
- The two transmitters could operate at different wavelengths.
In particular, he noted that two virus species which had a common host, and thus might be expected to encode similar proteins to cope with the common intracellular environment, differed dramatically in their (C+G) percentages (51.3% versus 38.4%; Table 1). The main hypothesis which will be developed here is that (C+G)% should be regarded as "secondary information", akin to wavelength in the transmission of radio messages. Different biological species "broadcast" their DNAs at different (C+G)% "wavelengths".
The selectionist dilemma is that of finding a selective force which could have driven the evolution of species differences in C+G percentages. It will be argued here that such a selective force exists, and is intimately linked to recombination (sex), a process which would have arisen very early in evolution.
- -Speciation can begin sympatrically
if the (C+G)% deviates from the species norm, perhaps
due to unopposed mutational biases.
- -Natural selection is constantly fine-tuning the (C+G)%
of DNA (through selection of fitter (C+G)% genotypes
which avoid recombination with the DNA of incipient species and other species in the same
environment).
- -Such a post-zygotic
isolating
mechanism provides a general basis
for speciation in all taxa; this will usually precede
the evolution of specific prezygotic isolating factors.
- -A change in environmental conditions which might favor new phenotypic characteristics
which had emerged together with the differences in (C+G)%,
or
- -Repeated "lucky" selection of gametes in small
populations (genetic drift).
The free single strands would then pair with complementary single stranded sequences at the tips of loops extruded from the duplex DNA of the homologous chromosome. This would initiate the recombination process. Crick's model made no explicit suggestion of ways DNA, the substrate of the enzymes mediating recombination, might mutate to favor recombination.
In 1972 the model was modified by Sobell, who proposed that the single stranded regions would be extruded as stem-loops. The stems would be formed by Watson-Crick base pairing and single-stranded loops at the tips of the stems would be available for pairing with homologs. Further elaborations of the model were introduced by Wagner and Radman (1975), and by Doyle (1978).
The first evidence for the model came from an unexpected source. In a certain bacterial plasmid the initiation of DNA synthesis is controlled by an RNA molecule transcribed from one strand of the DNA duplex. This RNA is regulated by an "antisense" RNA transcribed from the complementary strand. Tomizawa (1984) made detailed kinetic studies of the process by which the antisense RNA reacted with the complementary "sense" RNA derived from the other strand.
He concluded that the two molecules first interacted through the loops at the tips of stem-loop structures. If this "kissing" was successful (i.e. precise sequence complementarity was found), then the interaction was consummated by extending the pairing of sense and antisense strands. This pairing disrupted the stem-loop structure of the sense transcript and inhibited its role in initiating DNA synthesis (Fig. 1). In this case the purpose of the pairing was not to promote recombination, however the mechanism was consistent with the above stem-loop DNA recombination models. Evidence for a role of stem-loops in recombination between RNA genomes was presented by Romanova et al., (1986).
The stem-loop models have clear implications for the composition of DNA, since to be able to form stems, single stranded DNA should tend to obey Chargaff's rule (%A=%T; %C=%G). To a close approximation single stranded DNA does obey Chargaff's rule. In single strands of DNAs from all species studied there are approximately equal proportions of complementary bases, dinucleotides, trinucleotides and higher oligonucleotides (Nussinov, 1984; Pradhu, 1993; Forsdyke, 1995d). Indeed, in 1982 Nussinov deduced from this that stem-loops (inverted repeats) would be generated.
That at least some complementary oligonucleotide pairs have evolved in close proximity was suggested by studies using antibodies specific for stem-loop ("cruciform") DNA structures. Ward et al. (1990) assayed the number of cruciforms in DNA in intact cells to test the hypothesis that each cruciform might correspond to a site for the initiation of DNA synthesis ("replicon initiation"). They found far more sites than expected based on the calculated number of expected replicons (subunits of DNA which are independently replicated).When extensive sequences of DNA became available in the early 1990s, it was possible to look more precisely for the distribution of stem-loop potential in DNA (
Forsdyke, 1995b-e). The basic argument was that, if recombination is evolutionary advantageous, then mutations in DNA which favored recombination would be accepted.Two types
of favorable mutation were envisaged, mutations of the enzymes which mediate recombination, and mutations which improve the ability of DNA to act as a recombination substrate. The former mutations would have been localized to the regions of the corresponding genes. The latter mutations would have been dispersed. There would then have been a genome-wide evolutionary pressure on primary sequences promoting the acceptance of such mutations.Using appropriate computer programs it is possible to repeatedly test various folding patterns of a nucleic acid sequence, to arrive at an energetically most favorable configuration (Zuker, 1989). Using a moving sequence window, it is possible to scan along sequences to determine the distribution and extent of local stem-loop potential. The stability of stem-loops is a function both of base composition and of base order. Because of the greater strength of CG base pairing, a CG-rich sequence tends to have a more stable structure than an AT-rich sequence of the same length.
Indeed, the bases in a sequence might show poor complementarity, but if the few complementary pairs were CG pairs, the stability of the folded molecules might be quite high. Furthermore, as indicated earlier, base composition is a genome, or genome sector, "strategy", not a local "strategy" (Grantham et al., 1980; (Click Here)). Codons are an example of a local "strategy". Confidence that a calculated secondary structure is of local functional relevance is greater if it can be shown that the sequence has accepted mutations which enhance stem-loop stability by changing base order, rather than by changing base composition.
Using appropriate computer programs it is possible to randomize the sequence of bases in a nucleic acid molecule, thus disrupting the information in the primary sequence (base order), but not changing base composition. By comparing the folding of a "window" in a natural sequence with the average folding of numerous randomized versions of the same sequence window (Le & Maizel, 1989), it is possible to test whether there has been a local evolutionary pressure on base order promoting evolution of the potential to form stem-loops (Forsdyke, 1995b-e).
The difference between the minimum energy of folding of a natural sequence window (a function of base composition and base order), and the average of the corresponding minimum energies of folding of multiple randomized versions of the window (a function of base composition), is referred to as the "folding of randomized sequence difference" value (FORS-D value). The latter provides a measure of "base-order determined stem-loop potential". This is the component of the stem-loop potential which is of most local functional relevance, and closely corresponds to the "statistically significant" stem-loop potential of Le & Maizel (1989).
Long DNA sequences from a variety of species were scanned in this way, using a moving 200 nt window to ensure that only local stem-loop potential was evaluated. Base-order-determined stem-loop potential was found to be widely dispersed in DNA from different human chromosomes (including intergenic sequences) and from bacterial viruses (bacteriophage), bacteria (E. coli), and insects (Drosophila melanogaster; Forsdyke, 1995b-e). The potential was much more frequent than expected from the studies with anti-cruciform antibodies. The genome-wide pressure acting on base order to enhance stem-loop potential is referred to as " fold pressure" or " FORS-D pressure".
- Four DNA sequence differences can be regarded as presenting
consecutive barriers to recombination:
- -Differences in base composition-determined stem-loop potential (differences in
percentage C+G).
- -Differences in the base order of stems, or base insertions/deletions, which would
affect the proper positioning (register) of loops (Tomizawa,
1993).
- -Differences in base-order affecting the "kissing"
bases in loops.
- -Further differences in base order sufficient to trigger "fail-safe" mismatch repair enzymes leading to the "abortion" of recombining DNA molecules (Rayssiguier et al. 1989; Radman & Wagner, 1993).
The first row of arrows symbolizes the pressure to adopt a particular species-specific (C+G)% ("GC/AT pressure"). This provides the "base composition- determined" component of the stem-loop potential. A pressure acting to increase the ratio would increase the stability of stem-loops. A pressure acting to decrease the ratio would decrease the stability of stem-loops. The equilibrium between the two latter pressures is determined by interactions with other species in the same environment (see text).
The second row of arrows symbolizes a pressure to increase stem-loop formation by favouring the formation of inverted repeats with complementary oligonucleotide sequences ("fold pressure"). This provides the "base order-determined" component of the stem-loop potential and may be quantified as the FORS-D value.
The lower row of upward facing paired arrows symbolizes pressures for the encoding of specific functions in local regions of the genome (e.g. protein-mediated functions).
|
All forms of information share common features (Shannon, 1947). I here consider the transmission of information as radio-waves as a metaphor for the transmission of genetic information through time and space. A variety of observations, including recent work on the distribution of stem-loop potential in genomes (Forsdyke, 1995b-e), are shown consistent with a hypothesis which appears to throw new light on some fundamental questions in biology: The paper begins by considering the (C+G)% of a DNA molecule as "secondary information" which has been fine-tuned by natural selection to inhibit recombination with DNAs from other biological species (sections 2-5). It is then shown that stem-loop recombination models advanced in the 1970s can be adapted to explain how differences in (C+G)% might impede recombination (sections 6-7). Finally the ways different species balance the various evolutionary pressures on their genomes are explored (sections 8-11). |
|
 At the beginning of the 20th century the first radio transmitter came into
operation. A particular wave-band was chosen and messages were broadcast. The second radio transmitter to arise did not want to interfere with messages from the
first. There were three alternatives:
At the beginning of the 20th century the first radio transmitter came into
operation. A particular wave-band was chosen and messages were broadcast. The second radio transmitter to arise did not want to interfere with messages from the
first. There were three alternatives:|
As the number of transmitters increased the third alternative became the most feasible. Thus different transmitters often broadcast both synchronously and sympatrically. Operating on a second wavelength did not disadvantage the second transmitter. A message on one wavelength sounded much the same as on another wavelength. Thus the message was essentially wavelength-independent. This component of the radio signal can be regarded as "primary information". Information on wave-length is another component of the signal and can be regarded as "secondary information". DNA can also be considered to have both primary and secondary information components. Table 1 shows four columns of data adapted from a 1952 study by Wyatt on the base composition of DNA of various insect viruses. The first three columns demonstrate the famous "Chargaff ratios" (%A=%T; %G=%C; Chargaff, 1951). The data in these columns are species-invariant [i.e. are usually the same for all species] and led to the introduction of a general model for DNA in 1953 by Watson and Crick. Their model of DNA as a double helix was immediately successful in explaining many problems in genetics. The "primary information" in DNA, such as information coding for the sequence of a protein, was a characteristic of the sequence of the four bases. Individual DNA strands could act as templates permitting accurate replication of information. One strand could be used as a template to repair damage in the other. |
| VIRUS TYPE | Virus host | A/T | G/C | R/Y* | (C+G)% |
| Polyhedral.
. . . . |
P. dispar |
1.06 |
1.08 |
1.07 |
58.5 |
| L. monacha | 1.03 | 1.08 | 1.06 | 51.5 | |
| C. fumiferana | 1.03 | 1.09 | 1.06 | 51.3 | |
| M. americanum | 1.04 | 1.11 | 1.07 | 42.4 | |
| B. mori | 1.04 | 1.11 | 1.07 | 42.7 | |
| C. P. eurytheme | 1.08 | 1.11 | 1.09 | 42.5 | |
| N. sertifer | 1.07 | 1.09 | 1.07 | 37.4 | |
| Capsule . |
C. murinana |
1.05 |
1.11 |
1.07 |
37.4 |
| C. fumiferana | 1.01 | 1.12 | 1.05 | 34.8 | |
| * R = purine (A or G); Y = pyrimidine (C or T). | |||||
|
The initial success of the double helix model may have distracted attention from the data in Wyatt's fourth column. This shows that (C+G)% is a species characteristic. The ratio of the bases which engaged in strong Watson-Crick interactions (C and G), to the bases which engage in weak Watson-Crick interactions (A and T), is characteristic of a genome (or large genome sector; Sueoka, 1961; Muto & Osawa, 1987; Bernardi, 1989). In 1952 Wyatt was looking to (C+G)% for clues as to how DNA might carry its "primary information". He imagined that species with similar biological features might encode similar proteins and this might be reflected in similarities in base-ratios. He was forced to conclude that the (C+G)%:
|
|
Species with similar biological properties often encode similar proteins with similar primary sequences. It does not necessarily follow, however, that the DNA sequences which encode those proteins are as similar as the proteins. The genetic code, relating 61 nucleotide triplets in DNA to the 20 amino acids in proteins, is a redundant code. For some amino acids there is a choice of six possible codons (synonymous codons). Early sequencing studies showed that usage of alternative codons is
not random (Grosjean &
Fiers, 1982). In the "genome
hypothesis" (Click
Here) Grantham et al.(1980) pointed out that all genes in a genome tend to use
the same subset of codons. The needs of the genome seem to dominate codon
choice. The
codon subsets of a virus and its host are often different, even though they use the
same
translation machinery (Grantham et al.
1985). The same principle applies to sectored
vertebrate genomes. Thus, mammalian alpha-globin and beta-globin mRNAs share a common
cytoplasm, yet use different codon subsets. The corresponding genes are in the high and
low (C+G)% genomic sectors (isochores), respectively.
(C+G)% is a major factor determining
codon choice ( Possible explanations of species differences in (C+G)%
are at the heart of the debate between two schools. The " For example, CG bonds are less readily broken at
high temperatures. The selectionists have argued that high (C+G)%
genomes might have evolved as a response to the selective pressure of temperature. Indeed,
many contemporary organisms which survive at high temperatures do have CG-rich
DNA (Kagawa et al.
1984). However, the neutralists point out that some organisms living at
high temperatures have AT-rich DNA (for refs. see
Filipski, 1989, or Click Here
). Some neutralists now tend to
the extreme position that:
|

|
Sexual reproduction provides an opportunity for DNA from two individuals to recombine. The new gene combinations which result may be advantageous (Weismann, 1892). However, recombination can disrupt as well as create favorable gene combinations. For this reason, among others, it is difficult to accept the generation of advantageous gene combinations as the main driving force favoring the evolution of sex. There is currently much debate on the possible advantages of sexual reproduction compared with asexual reproduction. I here accept the postulate, most eloquently argued by the Bernsteins (1991), that recombination evolved primarily to correct DNA damage and mutations. Causes of the latter include DNA damage and replication errors. Organisms use recombination, among other methods, to maintain the integrity of their DNA. It is unlikely that a DNA molecule in one member of a species will be damaged or mutated at the same site as in the homologous DNA molecule in another (not closely related) member of the same species. One DNA molecule thus has the potential to act as a template for repair of, or to replace a defect in, another. Either a damaged base, or a mismatched base in a heteroduplex, can be dealt with. In the latter case, some mechanism to decide which is the correct strand is helpful. Organisms of different species may sometimes happen to have similar C+G percentages (" secondary information"; Table 1), but they invariably differ in base order ("primary information"). Thus, to act as a accurate template the DNA molecules must be from the same species. However, it is advantageous for organisms to avoid recombination with other members of their species whose DNA has deviated from the species norm (potential "incipient species"). The DNA of these members is no longer a reliable template for error detection and correction.Deviant members of a species can thus be seen as exerting a selection pressure for the evolution of mechanisms to monitor deviance from the species norm, and to prevent recombination if that deviance exceeds some unacceptable limit. The genomic deviations responsible for this impairment might precede the appearance of deviations giving rise to incompatibilities at the gene product level. Ideally the monitoring process would, in some way, first directly summate all the deviant aspects of a genome and then reject a genome which exceeded the limit. Alternatively, the monitoring process could assess some characteristic which would provide an indirect measure of genome deviance (e.g. percentage C+G). In the latter case there should be some explanation why difference species sometimes have very similar C+G percentages. One explanation would be that different genomes may differ locally in (C+G)%, but may maintain an overall similarity in (C+G)%. A more satisfying explanation is given later. |
|
Although a subject of much controversy, species can simply be defined as consisting of organisms which are successful at reproducing sexually with each other. Sexual species are reproductively isolated from other sexual species (Templeton, 1989; Coyne, 1992). Reproductive isolation may be due either to factors which impede fertilization (prezygotic factors: geographical, behavioral, anatomical), or to factors which act after fertilization (post-zygotic factors). Along lines first set out by
Dobzhansky (1936) and Muller (1939), current hypotheses postulate a major role for
incompatibilities between gene products. These incompatibilities might become greater as
species diverge, and, unless the products happened to influence fertility directly, would
tend to cause hybrid inviability
rather than infertility (Orr, 1995; Forsdyke,
1995a). In contrast, this paper focuses on
possible incompatibilities at the genomic level which would be manifest at meiosis
when parental genomes attempt to recombine, and
would cause hybrid infertility
[sterility].
The main postulates of the paper are that: Since to derive a benefit (DNA repair) from recombination an organism must use a homologous DNA molecule from another member of the same species, organisms of different (C+G)% "wavelengths" constitute a selective force driving the emergence of distinct species-specific C+G percentages. By making successful recombination contingent on this "secondary information" in DNA, the integrity of species DNA would be maintained. Thus, throughout evolutionary time each species would have fine-tuned the "wavelength" of its DNA to avoid interference from the DNA of other species. An individual organism from a subpopulation whose (C+G)% had begun to deviate from the species norm (potential incipient species) would usually fail to recombine with individuals from the majority population, and thus would not have the deviation corrected. The organism would also probably fail to recombine with other members from the subpopulation, because of their rarity. The subpopulation would thus be selected against unless rescued by: Wyatt (1952) noted that the range of C+G percentages in insect viruses was much wider than in other groups of organisms. This observation has been confirmed for viruses with other hosts (Bronson & Anderson, 1994). Viruses seem to have been most susceptible to "CG/AT pressure", which is the postulated evolutionary pressure driving a genome to adopt a particular (C+G)%. In the absence of special mechanisms to prevent coinfection (analogous to prezygotic isolation), the two species of DNA virus with the potential to coexist synchronously and sympatrically within cells of the C. fumiferana (Table 1), would have had every opportunity to recombine. Since they shared the challenges of a common environment ("ecological niche"), it is likely that they would have had some common proteins with similar sequences. If these sequences were similar at the DNA level then, in the absence of some barrier, recombination between the species would be possible (since recombination between DNA molecules is favored by sequence homology). In terms of the hypothesis advanced in this paper, there would thus have been a strong selective pressure on the DNA of these two viral species to arrive at distinct C+G percentages (analogous to post-zygotic isolation). A consequence of this would be that, even though they share a common translation apparatus, each species would have a distinct codon bias. Returning again to the somewhat imperfect radiowave metaphor, two transmitters which are close to each other (i.e. their transmitting ranges overlap) cannot broadcast simultaneously on the same wavelength. Similarly, two species which are biologically close to each other (i.e. prezygotic isolation may be imperfect), cannot "broadcast" on the same (C+G)% "wavelength" without interfering with each other. Two transmitters whose transmitting ranges do not overlap can broadcast at the same wavelength. Similarly, two biologic species which are reproductively isolated (through one or more prezygotic isolation factors), can "broadcast" their DNAs at the same (C+G)% "wavelength". If postzygotic isolation precedes prezygotic isolation, then initially reproductive isolation would require different C+G percentages; when prezygotic isolation was achieved, the two C+G percentages could converge, since there would no longer be a selective pressure for (C+G)% divergence. Thus, in modern species the process responsible for the initiation of reproductive isolation could have become disguised by subsequently developing prezygotic isolating factors which would ensure maintenance of reproductive isolation. How could differences in (C+G)% affect recombination? Many studies have been carried out on the molecular basis of recombination and many models have been advanced taking into account the results of such studies (Szostak et al. 1983; Holliday, 1990). The need to explain differences in (C+G)% has not been seen as requiring an explanation in terms of the models. However, some early "stem-loop" models, which postulate the involvement of stem-loops in single-stranded DNA, lend themselves most favorably to this purpose. A possible answer to the question will be arrived at here in two steps. First the growing evidence supporting stem-loop recombination models will be summarized. Then it will be shown how interactions between loops might be extremely sensitive to small differences in (C+G)%. |
|
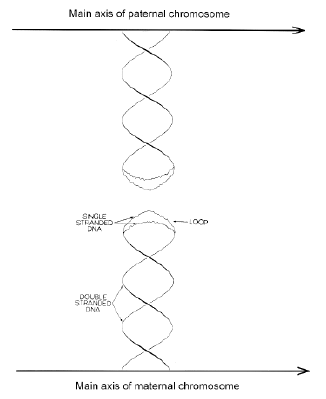
Critical to the reacceptance of early stem-loop recombination models are observations suggesting that the pairing of homologous chromosomes for recombination is not protein-based (i.e. does not require prior formation of a synaptonemal complex). Rather, chromosomal pairing is likely to be the result of a DNA sequence-based homology search (Kleckner et al., 1991; Hawley & Arbel, 1993; Kleckner & Weiner, 1993; Klein, 1994). In 1971 Crick advanced his "unpairing postulate" to explain how homologous chromosomes in diploid organisms might find each other. This invoked loops of duplex DNA at the tips of which complementary strands would unpair.
|
|
|
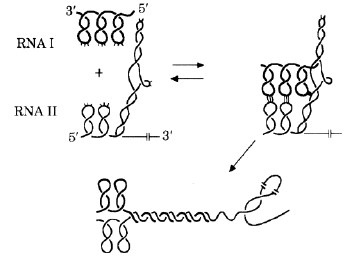
FIG. 1 . The stem-loop "kissing" model for the initiation of hybridization between two nucleic acid species (Tomizawa, 1984). Single stranded RNA I and its antisense transcript RNA II in plasmid ColE1 first interact transiently at the tips of stem-loops, mainly by conventional GC and AU base pairing. If this pairing is sufficiently stable, the pairing propagates progressively as the stem-loop structures unfold. The rate constant of association between loops is critical in determining the rate of subsequent hybridization. In the ColE1 system, the hybridization is required to regulate the initiation of DNA synthesis. In recombination systems the union would be further consummated by strand exchanges (Sobell, 1972; Wagner & Radman, 1975; Szostak et al., 1983). (This figure is adapted from Annual Reviews of Biochemistry, with the permission of Annual Reviews Inc; Eguchi et al., 1991). |
|
Unlike RNA, extrusion of stem-loops from duplex DNA requires torsional stress on the DNA duplex (supercoiling; Murchie et al., 1992). Thus, inhibition of supercoiling should impede recombination; indeed, this has been found (Wang et al., 1990). Furthermore, certain enzymes involved in recombination (endonucleases) work only with a supercoiled DNA substrate (Sung et al., 1993). The evidence from a variety of biological systems supporting the involvement of stem-loops in recombination has recently been summarized (Reed et al., 1994).
|
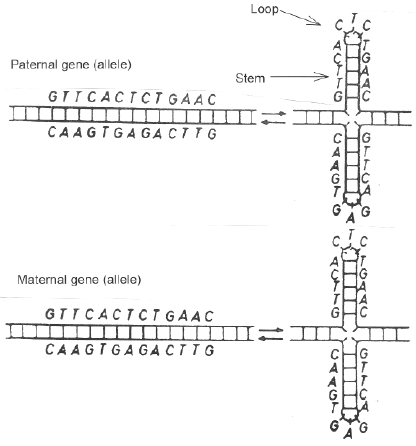
|
Tomizawa (1993) has concluded that the major role of the stem in a stem-loop structure is the proper positioning of the loop. This allows the unpaired bases in the loop to pair with those of an appropriately positioned complementary loop projecting from another nucleic acid. This "kissing" is rate-limiting in recombination. Base composition, a genomic rather than a local characteristic, is a major factor affecting the energetics of folding of a DNA segment (Forsdyke, 1995b-e; see also discussion of Fig. 3 in section 8). Small changes in this parameter would affect the looping pattern which a sequence could present for homology search. Furthermore, it would be more difficult to extrude loops from CG-rich DNA than from AT-rich DNA. Two sequences of different (C+G)% undergoing supercoiling in a common intracellular environment might extrude stem-loops at different times and to different extents. Thus, the pattern of loops presented by regions with quite similar sequences could be different and recombination could be impaired. To recombine, two homologous sequences should be equal both in the local parameter (base order-determined stem-loop potential) and the genomic parameter (base composition-determined stem-loop potential). |
|
It is likely that early biological evolution took place among "replicators" in an "RNA world" (Joyce & Orgel, 1993). At some point, either before or after the evolution of protein-encoding genes, the predominant nucleic acid became DNA. In the early nucleic acid world the ability to shuffle damaged segments so as to create new segment combinations should have been advantageous. Thus, if it could have evolved, it is likely that recombination would have evolved at an early stage. If the stem-loop model is applicable to this early world, then it can be imagined that primitive replicators which accepted mutations modifying their sequence to enhance the potential for stem-loop formation, would have had a selective advantage. To reap the benefits of efficient recombination a replicator would have had to exchange genetic segments with its own kind of replicator, not with other kinds. Since recombination with foreign replicators would have been disadvantageous, characteristics which prevent such recombination (e. g. a distinctly different C+G percentage) would have been favored by natural selection. Figure 2 symbolizes two early genome-wide pressures affecting the evolution of nucleic acids. The top row of downward-pointing arrows symbolizes a pressure affecting base-composition which would result in a particular (C+G)% ("CG/AT pressure"). The second row of downward-pointing arrows symbolizes a pressure on the primary sequence to accept mutations favoring an increase in stem-loop potential by changing base order (FORS-D or fold pressure). The two sets of arrows are pointing in the same direction. For present purposes the two pressures are not considered to conflict. FORS-D values can be considered as having been imposed on a CG/AT equilibrium which would have been arrived at independently. Many mutations currently regarded as "neutral" would in fact have been selected during the course of evolutionary adaptation to these pressures. |
|
|
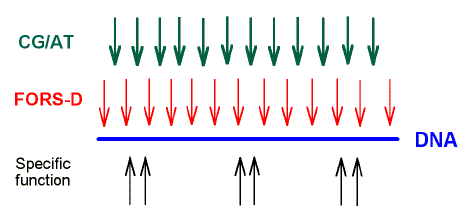
FIG. 2 . Genome-wide and local evolutionary forces acting on a genome. The two upper rows of dispersed downward-pointing arrows symbolize two genome-wide forces which influence the potential to form stem-loop structures.One way of accommodating the conflict between the pressures is to allow protein-encoding capacity to evolve in dispersed segments separated by regions (introns) where the evolution of stem-loop potential is less constrained. |
The upward pointing arrows in Figure 2 are in distinct regions,
symbolizing the later-evolving localized pressure for the encoding of specific function.
Here there is a conflict. A sequence required to encode a protein might not at the same
time be able locally to optimize its folding propensity. The conflict might have been meet
in three ways:
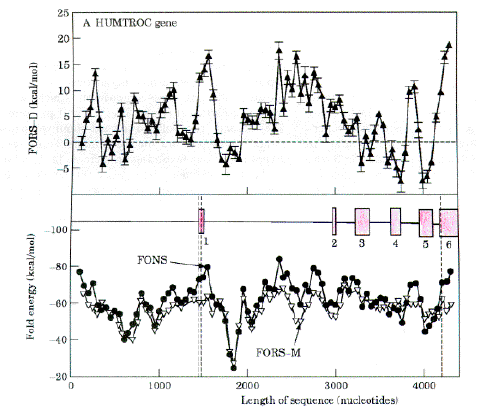
If the first two options were not sufficient, then only the third option would have been left. Thus, introns might correspond to parts of a gene where the constraints on the first two options were most severe. Introns would have allowed the interspersing of selectively advantageous stem-loops in coding regions of DNA. Evidence supporting this is presented elsewhere (Forsdyke, 1995b-e). As an example, Figure 3 (upper) shows FORS-D plots for the human troponin-c gene, which may have been under positive Darwinian evolutionary selection (Ohta, 1994). Negative FORS-D values are associated with certain exons. For exons which are not associated with negative FORS-D values, it can be assumed that it was possible to accommodate FORS-D pressure by the use of synonymous codons and conservative amino acids. Negative FORS-D values in parts of the first intron and 5' flank suggests functions for these regions, perhaps regulatory. Figure 3 (lower) shows that profiles for the folding of the natural sequence (FONS values) and the mean value for randomized sequences (FORS-M values), closely follow each other. This implies that the genome characteristic which controls the FORS-M value (base composition), is a major factor influencing the energetics of stem-loop formation (Forsdyke, 1995b-e). Once introns are removed, the cDNA product (not shown) has generally lower FORS-D values (average 2.31.9 kcal/mol) than the corresponding genomic segment shown in Figure 3 (average 4.40.7 kcal/mol).
|
|
|
- (i) non-synonymous codons, so that only conservative amino acid
changes are accepted.
- (ii) synonymous codons, so that only those which are translated most accurately are used (
FIG. 3. Comparison of fold energy minimization
values for windows in a human DNA segment containing the troponin-C gene, with arithmetical
mean values for multiple versions of each window repeatedly shuffled to randomize base
order before determining the fold energy minimization value (Forsdyke, 1995b-e). The first 4400 nt of the 4567 nt DNA sequence from GenBank file HUMTROC was divided into 85 windows of 200 nt, which overlapped each other by 150 nt. Secondary structure energy minimization values, determined using the program RNAFOLD (Zuker, 1989), were obtained for each window in the natural sequence (FONS values). Each 200 nt sequence was then subjected to 10 independent randomizations, and energy minimization values for each of the 10 randomized versions (FORS values) were determined using RNAFOLD. In A (lower) the mean fold value for each set of 10 randomized sequences (FORS-M value) is plotted with the corresponding FONS value. In A (upper) the differences between the FORS-M values and the corresponding FONS values are plotted (FORS-D values). [Comment: At this time directions of subtraction determined that high base order-dependent folding potential (FORS-D) was positive. Later the direction of subtraction was reversed to bring consistency with base composition-dependent folding potential (FORS-M; where high negative values indicate high folding potential). The top curve shows that base order-dependent folding is considerably constrained in exons 3, 4 and 5. DRF 2010] Each data-point is at the middle of its 200 nt window. Exon positions are shown as open boxes in A (lower). Vertical dashed lines in A indicate, from left to right, the beginning of exon 1, the beginning of the protein-encoding region, the end of the protein coding region, and the end of exon 6. |
Gilbert & Glynias (1993) postulate a correlation between exons and functional domains of proteins as part of their introns-early model. Failure to find the correlation has recently lead Stoltzfus and coworkers (1994) to embrace the introns-late viewpoint. The "introns-early" viewpoint presented here does not require a correlation of intron boundaries with some feature of protein structure. This is expected, since introns can occur both in protein-coding and in non-coding parts of a gene (Hawkins, 1988). Indeed, some genes encode "mRNAs" with no protein product, yet the genes have introns, just like protein-encoding genes (Brannan et al. 1990; Brockdorff et al. 1992). The viewpoint presented here suggests why introns were established, but does not explain their great variation in length. It is possible that this variation shifts the register between genes, and makes "kissing" interactions more difficult. |
9. (C+G)% AND PHYLOGENY
Similarities in (C+G)% between species are sometimes indicative of a close evolutionary (phylogenetic) relationship (Sueoka, 1961; Muto & Osawa, 1987). However, the arrows in Figure 2 indicate a potential conflict between CG/AT pressure and the later-evolving information to encode proteins (protein pressure). Figure 4 shows how CG/AT pressure, acting on a whole genome or large genome segment (Bernardi, 1989), might act independently of local protein pressures on the same genome or genome segment. The percentage identities of DNA and protein for various genes
in two organisms which have diverged from a common ancestor, are plotted against each
other. Exons of genes encoding a highly conserved protein (e.g. histone) might show
100%
identity at the protein level, but only 90% identity at the nucleotide level due to the
utilization of synonymous codons. The downward pointing arrow symbolizes this effect of CG/AT
pressure which tends to drive the (C+G)%
of the two species away
from each other (to prevent recombination). The environmental factor in this case is other close species, from which prezygotic isolation is of only limited effectiveness, and with which recombination must also be avoided. The conflicting directions of CG/AT pressure are in equilibrium in Figure 4 in the case of exons encoding proteins which show 75% identity between the two species. Thus, CG/AT pressure acts both to drive two species apart and together (away from other species). A phylogenetic relationship between the C+G percentages of the two species may still be evident. Data supporting this interpretation of percentage identity differences have recently been presented by Wolfe and Sharp (1993; although they do not offer this interpretation). They compared various homologous genes of mice and rats, and found that the cross-over point, when nucleotide identity equals amino acid identity, occurs at 93%. |
|
|
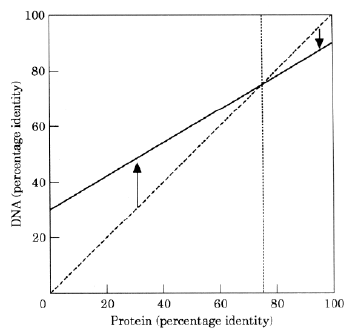
FIG. 4. Hypothetical relationship between degrees of nucleotide and amino acid identity for exons of two species which share a recent common ancestor. The diagonal dashed line indicates the relationship expected if corresponding nucleotide and amino acid sequences have changed equally during evolution. Exons of conserved proteins have small differences in amino acids (e.g. 100% identity between the two species), but CG/AT pressure (symbolized by the downward-pointing arrow) generates larger differences in nucleotides. This results in a sufficient difference in C+G percentage between the two species to inhibit recombination. Exons of poorly conserved proteins have large differences in amino acids (e.g. only 30% identity between the two species), but CG/AT pressure (symbolized by the upward-pointing arrow), working to prevent recombination with other species in the environment, conserves nucleotide identities. Thus, phylogenetic relationships between C+G percentages of the two species may still be evident. The vertical dashed line indicates the cross-over point (75% in this case) corresponding to exons of genes in which nucleotide and amino acid percentage identities are equal. |
However, whereas phylogenetically-unrelated species may sometimes have similar C+G percentages, phylogenetically-related species may differ markedly in (C+G)%. This is particularly evident in the case of the genomes of viruses which have the potential to share a common host cell where frequent opportunities for recombination might arise (e.g. C. fumiferana; Table 1). There is a 22% difference between the (C+G)% of two herpesviruses which are biologically very similar (Schachtel et al. 1991), and C+G percentage differences between biologically similar retroviruses are even greater (Bronson & Anderson, 1994). The latter authors do not relate this difference to the prevention of recombination, but suggest that it has arisen to allow viruses to occupy different "ecological niches" within a cell. |
10. MAJOR PRESSURES ON DNA ARE SELECTIVE
|
Major pressures affecting the evolution of DNA
are CG/AT pressure, and fold (FORS-D) pressure, as defined above. Two
other important pressures are protein pressure and translation pressure. Natural selection
results in the differential reproductive success of individuals with the functionally most
effective proteins, thus favoring genotypes which encode those proteins. This
inflexibility of amino acid sequence (protein pressure) places constraints on the use of :
Thus, in some circumstances, protein pressure might be manifest
as a positive correlation between non-synonymous and synonymous substitution rates in a
gene ( |
|
|
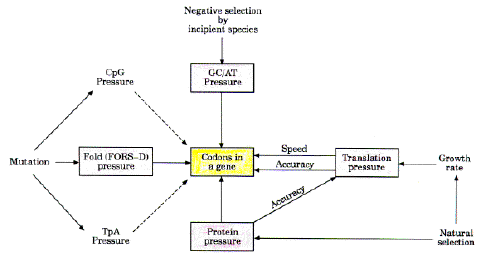
FIG. 5. Summary of pressures influencing the evolution of codons. The four major pressures are shown in boxes. For details please see text.
11. THE DOMINANCE OF THE GENOME
|
Salser in 1978, when analyzing the first globin mRNA sequences, challenged the current (and continuing) protein-centered view of sequence conservation during biological evolution:
Salser went on to show that base
substitutions predominate in mRNA loop regions, which are least likely to be involved in
maintaining the configuration of the folded RNA structure. This observation has since been
shown to apply to other RNAs (for refs. see Forsdyke,
1995e). |
This work was supported by the Medical Research Council of Canada.
REFERENCES
AKASHI, H. (1994). Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. J. Mol. Evol. 136, 927-935.
ALFF-STEINBERGER, C. (1987). Codon usage in Homo sapiens: Evidence for a coding pattern on the non-coding strand and evolutionary implications of dinucleotide discrimination. J. Theor. Biol. 124, 89-95.
BARRAI, I., SCAPOLI, C., GAMBARI, R. & BRUGNOLI, F. (1991). Frequencies of codons in histones, tubulins and fibrinogen: bias due to interference between transcriptional signals and protein function. J. Theor. Biol. 152, 405-426.
BERNARDI, G. & BERNARDI, G. (1986). Compositional constraints and genome evolution. J. Mol. Evol. 24, 1-11.
BERNARDI, G. (1989). The isochore organization of the human genome. Annu. Rev. Genet. 23, 637-661.
BERNSTEIN, C. & BERNSTEIN, H. (1991). Aging, Sex and DNA Repair. San Diego: Academic Press.
BIRD, A. P. (1980). DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8, 1499-1504.
BRANNAN, C. I., DEES, E. C., INGRAM, D. S. & TILGHMAN, S. M. (1990). The product of the H19 gene may function as an RNA. Mol. Cell. Biol. 10, 28-36.
BROCKDORFF, N., ASHWORTH, A., KAY, G. F., McCABE, V. M., NORRIS, D. P., COOPER, P. J., SWIFT, S. & RASTAN, S. (1992). The product of the mouse Xist gene is a 15 kb inactive X-specific transcript containing no conserved ORF and located in the nucleus. Cell 71, 515-526.
BRONSON, E. C. & ANDERSON, J. N. (1994). Nucleotide composition as a driving force in the evolution of retroviruses. J. Mol. Evol. 38, 506-532.
CHARGAFF, E. (1951). Structure and function of nucleic acids as cell constituents. Fed. Proc. 10, 654-659.
COYNE, J. A. (1992). Genetics and speciation. Nature 355, 511-515.
COX, E. C. & YANOFSKY, C. (1967). Altered base ratios in the DNA of an Echerichia coli mutator strain. Proc. Natl. Acad. Sci. USA 58, 1895-1902.
CRICK, F. (1971). General model for the chromosomes of higher organisms. Nature 234, 25-27.[See genesis of this paper in End Note Jan 2010 below]
DOBZHANSKY, T. (1936). Studies in hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics, 21, 113-135.
DOYLE, G. G. (1978). A general theory of chromosome pairing based on the palindromic DNA model of Sobell with modifications and amplification. J. Theor. Biol. 70, 171-184.
EGUCHI, Y., ITOH, T. & TOMIZAWA, J. (1991). Antisense RNA. Annu. Rev. Biochem. 60, 631-652.
FILIPSKI, J. (1990). Evolution of DNA sequence. Contributions of mutational bias and selection to the origin of chromosomal compartments. Adv. Mutagenesis Res. 2, 1-54.
FORSDYKE, D. R. (1995a). Fine-tuning of intracellular protein concentrations, a collective protein function involved in aneuploid lethality, sex-determination and speciation? J. Theor. Biol. 172, 335-345.
FORSDYKE, D. R. (1995b). A stem-loop "kissing" model for the initiation of recombination and the origin of introns. Mol. Biol. Evol. 12, 949-958.
FORSDYKE, D. R. (1995c). Conservation of stem-loop potential in introns of snake venom phospholipase A2 genes. An application of FORS-D analysis. Mol. Biol. Evol. 12, 1157-1165.
FORSDYKE, D. R. (1995d). Relative roles of primary sequence and (G+C)% in determining the hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J. Mol. Evol. 41, 573-581.
FORSDYKE, D. R. (1995e). Reciprocal relationship between stem-loop potential and substitution density in retroviral quasispecies under positive Darwinian selection. J. Mol. Evol. 41, 1022-1037.
GILBERT, W. & GLYNIAS, M. (1993). On the ancient nature of introns. Gene 135, 137-144.
GRANTHAM, R. (1980). Workings of the genetic code. Trends Biochem. Sci. 5, 327-331.(Click Here)
GRANTHAM, R., GREENLAND, T.,
LOUAIL, S., MOUCHIROUD, D., PRATO, J. L., GOUY, M. & GAUTIER, C. (1985). Molecular
evolution of viruses as seen by nucleic acid sequence study. Bull. Inst.
Past. 83,
95-145.
GROSJEAN, H. & FIERS, W. (1982). Preferential codon usage in prokaryotic genes: the
optimal codon-anticodon interaction energy and the selective codon usage in efficiently
expressed genes. Gene 18, 199-209.
HAWKINS, J. D. (1988). A survey of intron and exon lengths. Nucleic Acids
Res. 16,
9853-9905.
HAWLEY, R. S. & ARBEL, T. (1993). Yeast genetics and the fall of the classical view of
meiosis. Cell 72, 301-303.
HOLLIDAY, R. (1990). The history of heteroduplex DNA. BioEssays 12, 133-141.
IKEMURA, T. (1981). Correlation between the abundance of Echerichia coli transfer
RNAs and the occurrence of the respective codons in its protein genes. J. Mol.
Biol.
146, 1-21.
JOYCE, G. F. & ORGEL, L. E. (1993). Prospects for understanding the origin of the RNA
world. In: The RNA
World.
(Gesteland, R. F. & Atkins, J. F. eds.), pp. 1-25. New York: Cold Spring Harbour
Laboratory Press.
KAGAWA, Y., NOJIMA, H., NUKIWA, N., ISHIZUKA, M., NAKAJIMA, T., YASUHARA, Y., TANAKA, T.
& OSHIMA, T. (1984). High G+C content in the third letter of codons of an extreme
thermophile.
J. Biol. Chem. 259, 2956-2960.
KIMURA, M. (1989). The neutral theory of molecular evolution and the world view of the
neutralists. Genome 31, 24-31.
KLECKNER, N., PADMORE, R. & BISHOP, D. K. (1991). Meiotic chromosome metabolism: one
view. Cold Spring Harbour Symp. Quant. Biol. 56, 729-743.
KLECKNER, N. & WEINER, B. M. (1993). Potential advantages of unstable interactions for
pairing of chromosomes in meiotic, somatic and premeiotic cells. Cold Spring Harbour
Symp. Quant. Biol. 58, 553-565.
KLEIN, S. (1994). Choose your partner: chromosome pairing in yeast meiosis. BioEssays
16, 869-871.
KURLAND, C. G. (1993). Major codon preference: theme and variation. Biochem. Soc.
Trans. 21, 841-846.
LE, S-Y. & MAIZEL, J. V. (1989). A method for assessing the statistical significance
of RNA folding. J. Theor. Biol. 138, 495-510.
LI, W, H. & GRAUR, D. (1991). Fundamentals of Molecular Evolution. pp. 77, Sunderland, Mass: Sinauer
Associates.
MEZA, L., ARAYA, A., LEON, G., KRAUSKOPF, M., SIDDIQUI, M. A. & GAREL, J. P. (1977).
Specific alanine tRNA species associated with fibroin biosynthesis in the posterior
silk-gland of Bombyx mori.
FEBS. Lett.
77, 255-260.
MULLER, H. J. (1939). Reversibility in evolution considered from the standpoint of
genetics.
Biol. Rev. Camb. Philos. Soc. 14, 261-280.
MURCHIE, A. I. H., BOWATER, R., ABOUL-ELA, F. & LILLEY, D. M. J. (1992). Helix opening
transitions in supercoiled DNA.
Biochem. Biophys. Acta
1131, 1-15.
MUTO, A. & OSAWA, S. (1987). The guanine and cytosine content of genomic DNA and
bacterial evolution. Proc. Natl. Acad. Sci. USA 84, 166-169.
NICHOLS, B. P., BLUMENBERG, M. & YANOFSKY, C. (1981). Comparison of the nucleotide
sequence of trpA and sequences immediately beyond the trp operon of Klebsiella
aerogenes, Salmonella typhi, and Escherichia coli. Nucleic Acids
Res. Nucleic Acids
Res. Nucleic Acids
Res. Nucleic Acids
Res. 9,
1743- 1755.
NUSSINOV, R. (1982). Some indications for inverse DNA duplication. J. Theor.
Biol. 95,
783-793.
NUSSINOV. R. (1984). Doublet frequencies in evolutionarily distinct groups. Nucleic
Acids Res. 12, 1749-1763.
OHTA, T. (1994). Further examples of evolution by gene duplication revealed through DNA
sequence comparisons. Genetics 138, 1331-1337.
ORR, H. A. (1995). The population genetics of speciation: the evolution of hybrid
incompatibility. Genetics 139, 1805-1813.
PRADHU, V. V. (1993). Symmetry observations in long nucleotide sequences. Nucleic Acids
Res. 21, 2797-2800.
RADMAN, M. & WAGNER, R. (1993). Mismatch recognition in chromosomal interactions and
speciation. Chromosoma 102, 369-373.
RAYSSIGUIER, C., THALER, D. & RADMAN, M. (1989). The barrier to recombination between Echerichia
coli and Salmonella typhimurium is disrupted in mismatch-repair mutants.
Nature
342, 396-401.
REED, K. M., BEUKEBOOM, L. W., EICKBUSH, D. G. & WERREN, J. H. (1994). Junction
between repetitive DNAs on the PSR chromosome of Nasonia vitripennis: association
of palindromes with recombination. J. Mol. Evol. 38, 352-362.
ROMANOVA, L. I., BLINOV, V. M., TOLSKAYA, E. A., VIKTOROVA, E. G., KOLESNIKOVA, M. S.,
GUSEVA, E. A. & AGOL, V. I. (1986). The primary structure of crossover regions of
intertypic poliovirus recombinants: a model or recombination between RNA genomes.
Virology
155, 202-213.
SALSER, W. (1978). Globin mRNA sequences: analysis of base pairing and evolutionary
implications. Cold Spring Harb. Symp. Quant. Biol. 42, 985-1002.
SCHACHTEL, G. A., BUCHER, P., MORCARSKI, E. S., BLAISDELL, B. E. & KARLIN, S. (1991).
Evidence for selective evolution of codon usage in conserved amino acid segments of human
alphaherpesvirus proteins. J. Mol.
Evol. 33, 483-494.
SHANNON, C. E. (1948) The mathematical theory of communication. Bell Syst. Tech. J.
27, 397-423.
SHARP, P. M., STENICO, M., PEDEN. J. F. & LLOYD, A. T. (1993). Codon usage: mutational
bias, translation selection, or both? Biochem. Soc.
Trans. 21, 835-841.
SOBELL, H. M. (1972). Molecular mechanism for genetic recombination. Proc. Natl. Acad.
Sci. USA 69, 2483-2487.
STOLTZFUS, A., SPENCER, D. F., ZUKER, M., LOGSDON, J. M. & DOOLOTTLE, W. F. (1994).
Testing the exon theory of genes: the evidence from protein structure. Science 265,
202-207.
SUEOKA, N. (1961). Compositional correlation between deoxyribonucleic acid and protein.
Cold
Spring Harbor Symp. Quant. Biol. 26, 35-43.
SUNG, P., REYNOLDS, P., PRAKASH, L. & PRAKASH, S. (1993). Purification and
characterization of the Saccharomyces cerevisiae RAD1/RAD10 endonuclease.
J.
Biol. Chem. 268, 26391-26399.
SZOSTAK, J. W., ORR-WEAVER, T. L. & ROTHSTEIN, R. J. (1983). The double- strand-break
repair model for recombination. Cell
33, 25-35.
TEMPLETON, A. R. (1989). The meaning of species and speciation: a genetic perspective. In:
Speciation and its
Consequences.
(Otte, D. & Endler, J. A. eds), pp. 3-27, Sunderland, Mass: Sinauer Associates.
TOMIZAWA, J. (1984). Control of ColE1 plasmid replication: the process of binding of RNA I
to the primer transcript. Cell 38, 861-870.
TOMIZAWA, J. (1993). Evolution of functional structures of RNA. In: The RNA World.(Gesteland, R. F. & Atkins,
J. F. eds.), pp. 419-445, New York: Cold Spring Harbour Laboratory Press.
WAGNER, R. E. & RADMAN, M. (1975). A mechanism for initiation of genetic
recombination. Proc. Natl. Acad. Sci. USA 72, 3619-3622.
WANG. J. C., CARON, P. R. & KIM, R. A. (1990). The role of DNA topoisomerase in
recombination and genome stability: a double-edged sword? Cell 62, 403-406.
WARD, G. K., MCKENZIE, R., ZANNIS-HADJOPOULOS, M. & PRICE, G. B. (1990). The dynamic
distribution and quantification of DNA cruciforms in eukaryotic nuclei. Exp. Cell Res.
188, 235-246.
WATSON, J. D. & CRICK, F. H. C. (1953). Genetical implications of the structure of
deoxyribonucleic acid. Nature 171, 964-967.
WEISMANN, A. (1892). Essays
upon heredity and kindred biological problems. Vol. 2. Oxford: Clarendon Press.
WOLFE, K. H. & SHARP, P. M. (1993). Mammalian gene evolution: nucleotide sequence
divergence between mouse and rat. J. Mol.
Evol. 37, 441-456.
WYATT, G. R. (1952). The nucleic acids of some insect viruses. J. Gen. Physiol. 36,
201- 205.
ZUKER, M. (1989). Computer prediction of RNA secondary structure. Meth.
Enzym. 180,
262-289.
|
This paper postulated that viruses with the potential to occupy a common cytosol must differ in their base compositions in order to emerge as distinct species. This principle now appears to provide a theoretical under-pinning for the use of base compositions to classify viruses. In simple form, two highly conserved regions in a species are employed as PCR primers and the variable intermediate regions then become available for determination of base composition by ESI-MS (electrospray ionization and mass spectrometry). No sequence information (base order) is needed. If necessary several primer pairs can be used. Thus, given a patient sample, multiple pathogens can be independently identified with great precision. Indeed, "priming across broadly conserved regions provides taxonomic resolution at the species level" (Sempath et al. 2007a). Of particular relevance to the present paper is that, as expected from theory, the assay can provide an early warning of an emerging species (Sempath et al. 2007b). Sempath et al. (2007a) Rapid identification of emerging infectious agents using PCR and electrospray ionization mass spectrometry. Annals of the New York Academy of Sciences 1102, 109-120. Sempath et al. (2007b) Global surveillance of emerging influenze virus genotypes by mass spectrometry. PLOS One 2, issue 5, e489. |
|
Further scientific developments are considered in an End Note to a later paper Click Here. A recent biography of Crick (Olby 2009) contains much information on the genesis of Crick's "unpairing postulate" paper (1971; cited above). Olby concludes (p. 359) that "With the advent of the nucleosome, Crick's model of 1971 was now only of historical interest." Certainly, citations in the literature would seem to support this. They peaked in 1974 at 59 and then declined exponentially to zero in 1984. But Olby, among many others, appeared not to take account of the legend to Crick's Figure 3. I began to cite Crick's paper in the mid-1990s, and my citations, plus those of historians, have since sustained annual citations around 0 - 4. My interest in the paper arose from Crick's Figure 3 (slightly modified above in this web-page and not included in my original paper). In this context, the letter of transmission which Crick sent to the Editor of Nature on 3rd September 1971 is of considerable interest (Olby, page 352):
Thus, it would seem that Crick put considerable weight on the legend to Figure 3, his attention to nucleosomes being somewhat of a decoy in this respect. Olby R (2009) Francis Crick. Hunter of Life's Secrets. Cold Spring Harbor Laboratory Press. |
|
Having reviewed for PLOS Genetics two papers on base composition (GC%) by Hildebrand et al. (2010) and by Hershberg and Petrov (2010), Eduardo Rocha and Edward Feil announced (2010) that "we are facing a seismic shift of paradigm in molecular evolution." They produced a table of "variables historically proposed to explain GC variation in prokaryotes," among which was mention of the above paper. However, they took exception to it because "it does not explain why there are traces of pervasive selection for GC." No further explanation for their disdain was offered. Hershberg R & Petrov DA (2010) Evidence that mutation is universally biased towards AT in bacteria. PLOS Genetics 6, e1001115. Hildebrand F, Meyer A & Eyre-Walker A (2010) Evidence of selection upon genomic GC-content in bacteria. PLOS Genetics 6, e1001107. Rocha EPC & Feil EJ (2010) Mutational patterns cannot explain genome composition: are there any neutral sites in the genomes of bacteria? PLOS Genetics 6, e1001104.
|
|
Beautiful work on the role of nucleic structures in recombination between polioviruses (Romanova et al. 1986, see above; Tolskaya et al. 1987) has been confirmed and extended by Runckel et al. (2013). Furthermore, they provide strong evidence that GC%, which would tend to stabilize such structures, positively supports recombination. However, for poliovirus they were unable to support the idea that recombination preferentially occurs at gene boundaries, so tending to preserve intact genes. Runckel C, Westesson O, Andino R, DeRisi JL (2013) Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLOS Pathogens 9, e1003164. Tolskaya EA, Romanova LI, Blinov VM, Viktorova EG, Sinyakov AN et al. (1987) Studies on the recombination between RNA genomes of poliovirus. The primary structure and nonrandom distribution of crossover regions in the genomes of intertypic poliovirus recombinants. Virology 161, 54-61. |
Next: Thinking about Stem-Loops (1998) (Click Here)
Return to Bioinformatics Index (Click Here)
Return to Evolution Index (Click Here)
Return to HomePage (Click Here)
This page was established circa 1998 and was last edited 23 Nov 2014 by D. R. Forsdyke