Goals
Our goals are to improve the diagnosis, prognosis and treatment of patients with myeloid cancers, by studying their genetic and immune origins, including from the common age-associated condition called clonal hematopoiesis (CH or CHIP).
Research Directions
I) Translational and Transformative Myeloid Cancer Pathology
 Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoietic neoplasms, manifesting with dysplastic bone marrow failure, peripheral blood cytopenias and variable predisposition for transformation to acute myeloid leukemia (AML). Diagnosis is heavily reliant on non-specific, subjective morphology, particularly in early stages – karyotype results are most often normal, and few MDS-specific molecular assays currently exist. Diagnosis is often delayed, requiring serial invasive bone marrow investigations. Moreover, MDS primarily afflicts older adults and, without molecular targets and assays, treatments are mainly supportive (i.e. blood transfusions and growth factors). While some MDS patients respond to emerging agents, such as hypomethylating agents, it is not entirely clear how to predict treatment responses. With our aging demographics, MDS prevalence is expected to increase, along with demands for novel and more personalized treatment.
Myelodysplastic syndromes (MDS) are a heterogeneous group of clonal hematopoietic neoplasms, manifesting with dysplastic bone marrow failure, peripheral blood cytopenias and variable predisposition for transformation to acute myeloid leukemia (AML). Diagnosis is heavily reliant on non-specific, subjective morphology, particularly in early stages – karyotype results are most often normal, and few MDS-specific molecular assays currently exist. Diagnosis is often delayed, requiring serial invasive bone marrow investigations. Moreover, MDS primarily afflicts older adults and, without molecular targets and assays, treatments are mainly supportive (i.e. blood transfusions and growth factors). While some MDS patients respond to emerging agents, such as hypomethylating agents, it is not entirely clear how to predict treatment responses. With our aging demographics, MDS prevalence is expected to increase, along with demands for novel and more personalized treatment.
Recurring MDS-associated somatic mutations and gene expression signatures have recently been discovered that convey diagnostic and prognostic information not captured with current pathology platforms. In order to address these shortcomings, we have assembled clinically-rich MDS databases, bone marrow banks and clinical collaborators at Sunnybrook Health Sciences Centre and Kingston Health Sciences Centre, and access to bone marrow specimens from an NCIC/SWOG MDS clinical trial. Moreover, we have available at Queen’s University core next-generation sequencing (NGS) facilities, a CFI-funded NanoString nCounter gene expression analysis platform, and the histopathology resources of the Queen’s Laboratory for Molecular Pathology.
Specific Aims:
- Diagnostic: NGS may provide a more objective molecular test that can identify clonal versus reversible, non-clonal causes of dysplasia, leading to closer clinical monitoring and earlier identification of potentially targetable driver mutations. Together, these may lead to earlier and less invasive detection of MDS.
- Prognostic: The gene mutation panel and NanoString nCounter-based digital mRNA expression assay may contribute to refined clinical risk classification of MDS patients, and may eventual help to tailor patient/risk-specific treatment.
- Predictive: The mutation panel includes genes involved in epigenetic regulation and this may help predict which patients will benefit from hypomethylating agents and which patients should be spared unnecessary side effects.
Our related research experience with NGS has been instrumental in the early adoption of clinical myeloid NGS testing at Kingston Health Sciences Centre (KHSC), where we have subsequently demonstrated its clinical utility to inform diagnosis, prognosis and clinical management.
As will be discussed in subsequent sections, our research focus is now expanding to the detection of "pre-cancerous" clonal hematopoiesis, setting the stage for earlier myeloid cancer detection and perhaps intervention/prevention.

II) The Immune Environment of CHIP and MDS – Impacts of TET2 and DNMT3A Mutations
During MD/PhD training at the Terry Fox Laboratory with Gerald Krystal, I contributed to seminal studies in Immunity demonstrating Ship1 as a master regulator of macrophage polarization. Macrophages in the Ship1-deficient mouse model of MDS/MPN were profoundly “M2-skewed”, including constitutively high expression of arginase 1 (Arg1) associated with chronic inflammation adaptation and tumour immune evasion.
During Hematopathology residency training at U of T with Rena Buckstein and Richard Wells, and my initial practice at Queen’s, we translated these murine findings to humans, demonstrating a similar pathogenic mechanism in early MDS and MDS/MPN driven by mutations in epigenetic regulators, TET2 and DNMT3A. Our work on myeloid NGS has implicated these and other mutant genes in clinical outcomes for MDS and MDS/MPN patients and was instrumental in the early clinical adoption of myeloid NGS at Kingston Health Sciences Centre.
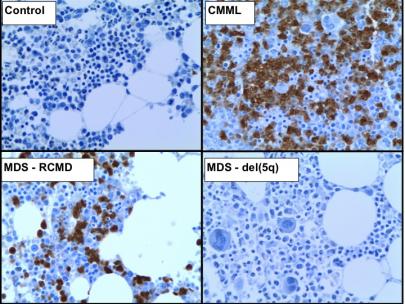
In this process, we noted how inactivating TET2 mutations were early drivers of myeloid cancers and skewed hematopoietic differentiation to the monocytic lineage. Therefore, my lab acquired and established the Cre-Lox system of murine hematopoietic Tet2-knockout, to study the effects of what seemed a highly clinically relevant and important regulator of macrophages.
In work that was slightly preceded by a paper published in Nature, yet contemporary with similar studies in Science and NEJM, we showed that Tet2 restrains inflammatory gene expression in macrophages. We then demonstrated that the inflammatory environment created by Tet2-mutant progeny favours the expansion of Tet2- mutant hematopoietic stem/progenitor cells (HSPC) at the expense of normal HSPC, through resistance to apoptosis. This finding was subsequently confirmed and extended by another group in Cell Stem Cell, providing insight into the origins of myeloid neoplasms.
With this knowledge, the bench to bedside nature of my program, and with the collaboration of Rena Buckstein and others at Sunnybrook and Baycrest (Toronto), we turned our attention to human clonal hematopoiesis (CH or CHIP). In work that was featured as a 2018 Highlight of ASH, in ASH Clinical News, and published at Blood Advances, we showed for the first time that human CH (dominated by mutations in DNMT3A and TET2) is associated with increased serum levels of inflammatory cytokines, like IL-6 and TNFα, increased frailty, increased and novel comorbidities (possibly including thyroid and gastrointestinal disease). CH is an established risk factor for atherosclerotic cardiovascular disease; however, we have also identified TET2-deficient blood cells as novel risk factors and mediators of the inflammatory cardio-pulmonary disease known as pulmonary arterial hypertension (Circulation, 2020).
This expertise at the intersection of inflammation/CH has been recognized by several invited review/methodological articles/editorials, my co-chairing of the first open CH abstract session at ASH 2018, and invitations to peer-review CH article submitted to journals like NEJM and Nature.
There are exciting possibilities regarding mitigation of leukemia risk and comorbid inflammatory diseases by targeting CH inflammatory processes. However, the focus in CH to this point has been macrophage-biased and mainly informed by murine studies. We have identified an RNA-seq signature of CH in bulk human peripheral blood based on deregulated neutrophil-related genes and functional neutrophil changes in Tet2-ko mice.
The next steps of our plan are to broaden the understanding of the effects of CH mutations in other cell types (esp. human), including at the single-cell level, and to explore the therapeutic potential of targeting associated inflammation.
- Address the lack of basic knowledge regarding the role of TET2 and DNMT3A and related cancer-driver mutations in murine and human myelomonocytic cell function - monocytes, macrophages and granulocytes (neutrophils) - including at single cell and organismal levels.
- Determine how aging and inflammatory processes contribute to the favoured expansion of TET2- and DNMT3A-mutant CHIP clones, and the potential for therapeutic intervention, using model systems.
III) Clonal Hematopoiesis - Novel Research and Clinical Directions
Early cancer detection is critical to improving health outcomes and cancer survival in the Canadian population. Detection of hematologic cancers, such as myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS), acute myeloid leukemia (AML), and diffuse large B-cell lymphoma (DLBCL) is currently dependent on invasive sampling of bone marrow or lymphoid tissue and specialized testing available at tertiary centres. As a result, most patients present late with progressed disease, where treatment options are costly, mostly non-targeted, and often ineffective.
With our team and collaborators, we have led major discoveries and programs to improve early cancer genomic studies and biomarker development. Myeloid cancers can now be detected at their earliest genetic stage, termed age-related clonal hematopoiesis (ARCH) or clonal hematopoiesis of indeterminate potential (CHIP). For simplicity, the term “CH” is used to refer to all clonal hematopoiesis. Importantly, CH occurs long before the conventionally-defined clinical presentation of these neoplasms as hematopoietic stem and progenitor (HSPC) cells commonly acquire genetic mutations or mosaic chromosomal abnormalities (mCAs) with advancing age, particularly in the epigenetic regulatory genes, DNMT3A and TET2. These mutations confer a competitive advantage such that progeny of these mutant stem cells expand in blood and become detectable by DNA sequencing. CH is found in at least 15-20% of adults >60 years. CH increases the risk of, and is a precursor to, hematologic cancers and can, therefore, be used for surveillance and risk prediction. However, with an annualized risk of 0.5-1%, most people with CH will not progress to cancer. We wish to improve on this and study how best to optimize biomarkers to find those at greatest risk for MDS, AML and other hematologic cancers.
We have also reported that these mutant blood cells infiltrate various body tissues, contribute to and accumulate in an inflammatory environment, and exacerbate a substantial and growing list of other diseases, including heart, lung and infectious diseases. We are exploring mechanisms and related ways to target CH mutant cells as a means of preventing blood cancer and ensuring healthier aging.
Specific Aims:
-
We will build on our strong CH foundation to detect and characterize the evolution of other myeloid cancers and lymphoid cancers. We will identify hematologically-healthy participants in longitudinal Canadian population health studies who develop incident hematologic cancers, along with matched controls. Through our pathology network we will access matched blood and hematologic cancer specimens that will provide a unique window on cancer evolution from CH. We will integrate temporal multi-omic and clinical information to provide a robust platform to study aging-related changes in the hematopoietic system in health and disease, and models to improve our current pre-cancer transformation model accuracy. We will partner with patient groups and primary care providers to identify barriers and opportunities related to CH testing.
- By exploiting the dependency and contribution of CH clones to aging- and inflammation-related changes in the host environment, we will try to identify new therapeutic strategies to target CH clones, prevent cancer development and co-morbidity.
- Using population and disease cohorts, and model systems, we will strengthen our understanding of and identify new diseases and risks associated with CH, including non-myeloid cancer, cardio-pulmonary, kidney, infectious and neurodegenerative diseases, and the potential for CH-targeted interventions.